
Abstract
Introduction
Age-related macular degeneration (AMD) is the main cause of blindness, which threatens the quality of life of older adults.1-3 Thus far, the pathogenesis of dry AMD is still unclear, and its emergence is related to various factors such as age, genetics, metabolism, environment, diet, and underlying systemic diseases. 4 Both animal model experiments and clinical studies involving age-related macular degeneration have shown that oxidative stress in the retinal pigment epithelial (RPE) is a key risk factor for the pathogenesis and progression of AMD. 5 Atrophic changes in the epithelium of the retinal pigment are key features of AMD progression. Aging and accumulated oxidative stress can trigger dysfunction of retinal pigment cells and eventually lead to cell death. 6 Targeting oxidative damage in RPE cells may be an effective strategy to improve early AMD. 7
Nuclear transcription factor E2-related factor 2 (Nrf2) is a transcription factor that senses the redox state and that is involved in various diseases, including AMD. 8 Nrf2 can be observed in the cytoplasm as a dimer formed with Kelch-like ECH-associated protein 1 (Keap1); however, when subjected to stimuli such as oxidative stress, it will dissociate from Keap1 and be transferred to the nucleus, where it binds to ARE and regulates the expression of ARE-mediated phase II detoxification and antioxidant enzymes, including heme oxygenase-1 (HO-1), glutamate-cysteine ligase (GCL), and NAD(P)H: quinone oxidoreductase (NQO1), 9 among which HO-1 is an important antioxidant enzyme that reduces cell damage. 10 The upregulation of Nrf2 protects retinal cells from oxidative damage and rescues the function of the retina.11-13 Recent studies have demonstrated that abundant bioactive compounds from natural products can activate the Nrf2/HO-1 signaling pathway to regulate the traumatic and stressed status of the retina. 14
Pachymic acid (PA) is extracted from the traditional Chinese medicinal herb Poria cocos, which belongs to the Lancaster-8-ene triterpene. PA exerts antitumor, anti-inflammatory, antioxidant, sedative, and hypnotic effects. 15 Studies have shown that PA can repair intestinal mucosal damage and reduce inflammatory responses in mice. In addition, PA protects the kidneys by activating the Nrf2/HO-1 pathway and inhibiting inflammation.11,16 Ming Mu Di Huang Wan (MMDH) pill restores the activity of antioxidant enzymes by activating Keap1/Nrf2/HO-1 signaling, which reduces cellular reactive oxygen species (ROS) levels and increases retinal pigment epithelial cell activity. 12 Goji berry, rehmania, and peony (the 3 main herbs in MMDH pills) can reduce oxidative stress and apoptosis in H2O2-induced RPE cells, 13 whether PA can prevent dry AMD remains unknown. Sodium iodate (SI) can induce RPE cell damage similar to the state of AMD, and H2O2 can induce RPE cell damage in vitro. 17 In this study, the anti-oxidative damage effect of PA on the retina was investigated through in vivo and in vitro experiments. We observed that PA prevents retinal damage from H2O2 in vitro or SI administration in vivo by activating the Nrf2/HO-1 signaling pathway, which inhibits ROS production and reduces apoptosis.
Materials and Methods
Agents and Cell Lines
PA (purity ≥98%, PU0510-0025) was purchased from Push Biotechnology. The PA used for the animal experiments was purchased from Nanjing Jin Yibai Biological Technology Co., Ltd. The SI used on the animals was purchased from Shanghai Macklin Biochemical Co., Ltd. The hydrogen peroxide solution was purchased from Guangdong Hengjian Pharmaceutical Co., Ltd Antibodies against anti-Nrf2 (16396-1-AP), anti-cleaved caspase-3, anti-HO-1, anti-Bcl-2, and anti-Bax were purchased from Proteintech. ML385 and ZnPP were purchased from AbMole Bioscience, and the human RPE cell line (ARPE-19) was obtained from the American Typical Culture Collection (ATCC, Manassas, VA, USA).
Cells were cultured in a complete medium (DMEM medium + 10% FBS + 1% antibiotics) at 37 °C in a humidified atmosphere of 5% CO2.
Cell Viability Assay
An MTT kit (Sevier G4101-1000T) was used to assess the cell viability in the different groups. ARPE-19 cells were seeded in 96-well plates for 24 h with 2 × 104 cells in each well and then stimulated with H2O2 (100-800 μmol/L) for 24 h. As for the PA-treated group, cells were pretreated with PA of different concentrations (10, 20, 40, and 80 μmol/L) before stimulation with H2O2 (400 μmol/L) for 24 h. In the end, the cells were incubated with MTT (20 μL) for 4 h, and the supernatant was removed afterward and lysed by DMSO. The absorbance of MTT in cells was measured at 570 nm. ZnPP or ML385 was then used to confirm the effect of PA on the cells.
Flow Cytometry
The cells of each group were collected and stained with annexin V/PI after different treatments (Yeasen Biotechnology Co., Ltd, Shanghai). The fluorescence was measured using flow cytometry, and the apoptosis rate was calculated based on the status of co-staining. All procedures were performed according to the manufacturer's instructions.
Mitochondrial Membrane Potential (MMP) Measurement
The MMP was measured using a JC-1 fluorescent probe (Servicebio) to measure the potential damage to mitochondria. After different treatments, RPE cells were subjected to 1 mL of JC-1 solution, incubated for 30 min at 37 °C, and protected from light. The ratio of red to green fluorescence was determined using fluorescence microscopy.
Determine the Intracellular ROS Level
DCFH-DA (2,7-dichlorofluorescein diacetate, Beyotime) was used to determine the level of intracellular ROS. RPE cells of each group were incubated with a 10 μM detecting compound for 30 min at 37 °C and protected from light. Subsequently, the levels of ROS in the ARPE cells were observed under a fluorescence microscope (OLYMPUS).
Western Blot
The cells of different groups were lysed, the proteins were extracted using RIPA (Best Bio) buffer and phenylmethylsulfonyl fluoride (PMSF), and their concentrations were determined using the BCA Protein Assay Kit (Beyotime). After electrophoresis and the transfer of proteins onto a blotting membrane, the blotting membrane was incubated with the primary antibody overnight at 4 °C. The excess primary antibody was washed off and incubated with the secondary antibody for 1 h. The intensity representing the expression levels of the proteins was measured using the ImageJ software. The following primary antibodies were used: anti-HO-1, anti-Nrf2 (Proteintech 16396-1-AP), and anti-cleaved caspase-3, Bax, and Bcl-2.
Animal
Twenty-four healthy male C57BL/6J mice (body weight 20-25 g) were purchased from Beijing Xinogenetic Co., Ltd (SCXK (Jing) 2022-0006). The mice had access to standard food and water at a room temperature of ∼ 22 °C to 24 °C with 12 h light/dark cycles. The animals were allowed to adapt for at least 1 week before the experiment. All procedures followed the guidelines of the Ethics Committee of the Liyang Traditional Chinese Medicine Hospital (No. 2023LY-1-20-01).
Twenty-four mice were randomly divided into 3 groups: (1) control group (n = 8): intraperitoneal injection of normal saline (NS) containing 0.1% DMSO once daily for 1 week, then tail intravenous injection of NS without SI once, and continued intraperitoneal injection of NS containing 0.1% DMSO once daily until the mice were euthanized. (2) Si group (n = 8): NS containing 0.1% DMSO was administered intraperitoneally once daily for 1 week, followed by tail intravenous administration of NS containing SI (40 mg/kg) once, and continued intraperitoneal administration of NS containing 0.1% DMSO once daily until the mice were euthanized. (3) SI + PA group (n = 8): intraperitoneal administration of NS containing 0.1% DMSO and PA (10 mg/kg) once daily for 1 week, followed by intravenous administration of NS containing SI (40 mg/kg) once daily, and then intraperitoneal administration of NS containing 0.1% DMSO and PA (10 mg/kg) once daily for 4 weeks until the mice were euthanized.
Micron IV Imaging
On day 28 of treatment, the mice were anesthetized with an intraperitoneal injection of tribromoethanol (400 mg/kg). Then, the retinas of 5 mice (both eyes) from each group were observed using a Micron IV retinal imaging camera system. The mice were subjected to optical coherence tomography (OCT) to observe the retinal morphology, and retinal thickness was measured using the ImageJ software.
Hematoxylin and Eosin (H&E) and Terminal dUTP Notched End Labeling (TUNEL) Staining
On day 28 after SI injection, the eyes of mice were freshly harvested and fixed overnight with eyeball fixative solution, dewaxed vertically (5 μm thick), then stained with H&E. Pathological changes were observed using light microscopy, and the retinal layer thickness was measured using the ImageJ software.
Apoptosis in the mouse retina was detected using an in situ cell death detection kit (Servicebio). Samples fixed in 4% paraformaldehyde were infiltrated with diluted Triton X-100 sodium citrate buffer for 2 min, and subjected to TUNEL staining and visualized with a diaminobenzidine (DAB) kit. The nuclei were counterstained with hematoxylin. The total number of apoptotic cells was counted as nuclei stained with TUNEL in 3 randomly selected fields of view for each tissue. The apoptosis rate was represented as the ratio of TUNEL-positive nuclei to the total nuclei/field of view.
Immunofluorescence
The fixed-section samples were permeabilized for 30 min and blocked (3% BSA) for 1 h at room temperature. A specific primary antibody was added and incubated overnight at 4 °C with anti-Nrf2 and HO-1 (Servicebio). The samples were then recovered at 37 °C for 1 h. The secondary antibodies were incubated for 1 h after washing off the excess primary antibody, and the nuclei were stained with DAPI for an additional 10 min. The specific fluorescence was observed using a fluorescence microscope (Nikon Eclipse C1, Tokyo, Japan).
Detection of SOD, CAT, and GSH-Px
The blood samples of mice from different groups were collected by eye removal into the centrifuge tube and centrifuged at 4000 r/min for 10 min at 4 °C after resting at room temperature for 1 h to collect serum samples. RPE cells were lysed by ultrasound and centrifuged to collect the supernatant according to the manufacturer's instructions.
After the mice were euthanized, a certain amount of tissue sample was added to cold PBS and homogenized in an ice bath, followed by centrifugation at 4 °C for 10 min. A BCA kit (Beyotime) was used to detect the protein concentration in the supernatant. Subsequently, the activities of GSH-Px (ml011256-J), catalase (CAT, ml095231-J), and superoxide dismutase (SOD, ml095231-J) were measured using the corresponding biochemical kits purchased from Shanghai Enzyme-linked Biotechnology Co., Ltd.
Statistics
The GraphPad Prism 9.0 statistical software (La Jolla, CA, USA) was used for data analysis. The data were expressed as the mean ± SD for a minimum of 3 independent experiments. One-way analysis of variance (ANOVA) and the least significant difference test were used to analyze significant differences in multiple and single comparisons, respectively. Statistical significance was set at P < .05.
Results
PA Improved Cell Viability Induced by Hydrogen Peroxide
First, we used MTT to measure the effect of different concentrations of the incubating cells with different concentrations of H2O2 (100, 200, 400, and 800 μmol/L) on cell viability for 24 h. The results showed that when the concentration of H2O2 reached 400 μmol/L, the cell viability decreased significantly by ∼50% compared with that in the blank control group (P < .05) (Figure 1a). H2O2 with a concentration of 400 μM was selected for the next series of experiments. Next, we examined whether PA (10, 20, 40, or 80 μmol/L) alone had adverse effects on ARPE-19 cells. As we can see, the cell activity was more than 90% when the PA concentration was 20 μmol/L, and the cytotoxicity could be ignored. Compared with that in the group with a PA concentration of 0 μmol/L, the cell viability of the group with a PA concentration of 80 μmol/L by more than 50% (P < .05) (Figure 1b), which could mean that the drug becomes toxic at higher concentrations. Therefore, this concentration was excluded in follow-up studies. Next, we evaluated whether pretreatment with PA mitigates the decrease in cell viability caused by H2O2. ARPE-19 cells were pretreated with PA for 2 h and stimulated with H2O2 (400 μmol/L) for 24 h. As shown in Figure 1c, 20 μmol/L of PA pretreatment for 2 h significantly eliminated (91.96%) (P < .05) the decreased cell viability induced by H2O2 induction (54.65%). Compared with that in the H2O2 (400 μmol/L) + PA (20 μmol/L) group (93.60%), the cell viability was significantly decreased after adding the Nrf2/HO-1 inhibitor (ML385: 70.19%; ZnPP: 64.73%) (Figure 1d). These results suggest that pretreatment with PA effectively mitigates the mortality caused by H2O2 through the Nrf2/HO-1 pathway.

PA improved cell viability in ARPE-19 cells induced by hydrogen peroxide. (A) Cell viability of ARPE-19 cells treated with different concentrations of H2O2 for 24 h. (B) Cell viability of ARPE-19 cells treated with different concentrations of PA for 24 h. (C) Cell viability of ARPE-19 cells pretreated with PA (10, 20, or 40 μmol/L) for 2 h, followed by H2O2 (400 μmol/L) for 24 h. (D) Cell viability of pretreatment of cells with or without ZnPP, ML385 (10 μmol/L) PA (20 μmol/L) for 2 h, followed by H2O2 (400 μmol/L) for 24 h. The viability was detected by MTT. The experiment was repeated 3 times (*P < .05 vs control, #P < .05 vs H2O2-treated group, $P < .05 vs PA 20 μmol/L).
PA Reduced the Cell Apoptosis of Retinal Cells Caused by Oxidative Stress
As shown in Figure 2a, after annexin V/PI staining, apoptosis was detected using flow cytometry and analyzed using a specific application. Compared with that in the control group (3.55%), the apoptosis rate of the H2O2 group was significantly higher (8.84%) (P < .05). However, apoptosis was significantly reduced after the addition of PA (4.71%) (P < .05), and this effect was reversed by treatment with Nrf2/HO-1 inhibitors (ML385: 5.47%; ZnPP: 5.61%) (Figure 2a and b). The apoptosis-inhibiting effect (P < .05) of PA on SI-treated mice was detected using TUNEL staining, which showed the same results as those obtained in vitro (Figure 2c and d).

PA reduces apoptosis of retinal cells induced by oxidative stress. (A, B) Apoptosis was analyzed using annexin V/PI staining in each group of ARPE-19 cells. (C) TUNEL-positive cells were detected in the retinal sections (green) (magnification 100 ×) (5 mice/10 eyes per group). (D) Quantitative display of TUNEL detection of apoptosis of retinal cells in vivo (5 mice/10 eyes per group). (E, F) Expression of apoptosis-associated proteins by WB. Experiments were repeated 3 times (*P < .05 vs control, #P < .05 vs H2O2-treated group, $P < .05 vs PA 20 μmol/L, ^P < .05 vs sham, +P < .05 vs SI-treated group).
Furthermore, we investigated the expression of apoptosis-associated proteins in ARPE cells using western blotting. The expression of Bax and cleaved caspase-3 was upregulated (P < .05), and that of Bcl-2 was down-regulated (P < .05) in the H2O2 group cells (Figure 2e and f). PA treatment significantly reversed the expression of these proteins, which was blocked in the Nrf2/HO-1 inhibitor-treated groups, indicating that PA may exert a protective effect on RPE cells from H2O2 stimulation by inhibiting apoptosis through the Nrf2/HO-1 pathway.
PA Reduced Oxidative Stress Levels in Cellular and Mouse Models of Oxidative Damage
Next, we used a specified probe, DCFH-DA, to evaluate the effect of PA on ROS production in the RPE after H2O2 treatment. The ROS levels in the H2O2 group (8.81%) were significantly higher (P < .05) than those in the control group (0.56%). After adding PA (20 μM) (0.97%), the ROS levels decreased significantly (P < .05), and this effect was reversed by Nrf2/HO-1 inhibitors (ML385: 6.85%; ZnPP: 7.36%) (P < .05) (Figure 3a and b). We used the JC-1 probe to evaluate the state of MMP in each group (Figure 3c and d). MMP decreased (P < .05) after H2O2 stimulation (0.612%), and PA pretreatment significantly prevented (5.891%) (P < .05) the decline. We tested the activity of SOD, CAT, and GSH-Px in vivo and observed that PA treatment significantly restored (P < .05) the SI-induced decrease in SOD, GSH-Px, and CAT levels in the mouse retina (Figure 3e to g). These results showed that PA reduces retinal oxidative damage.

PA reduced oxidative stress levels in cellular and mouse models of oxidative damage. (A, B) Intracellular ROS levels detected by DCFH-DA. (C, D) MMP in cells with different treatments by JC-1 staining. (E-G) In vivo, SOD, CAT, and GSH-Px activity (5 mice per group). The experiments were repeated 3 times (*P < .05 vs control, #P < .05 vs SI-treated group,$P < .05 vs PA 20 μmol/L, ^P < .05 vs sham, +P < .05 vs SI-treated group).
The Protective Effect of PA on the Structure of the Retina
The effect of PA on SI-induced retinal damage was detected using optical coherence tomography. We observed that PA significantly reduced (P < .05) the thinning of the retina induced by SI treatment, and HE staining showed that the epithelial monolayer was disrupted after SI treatment; RPE cells exhibited a circular degenerative phenotype, major loss of RPE cells, aberration and thinning of ONL, and disturbance of INL (Figure 4a to d), while PA pretreatment preserved the retinal layers, including RPE, the cell densities of INL and ONL, and photoreceptors, reduced thinning of the retina, and the number of deposits (Figure 4e to h).
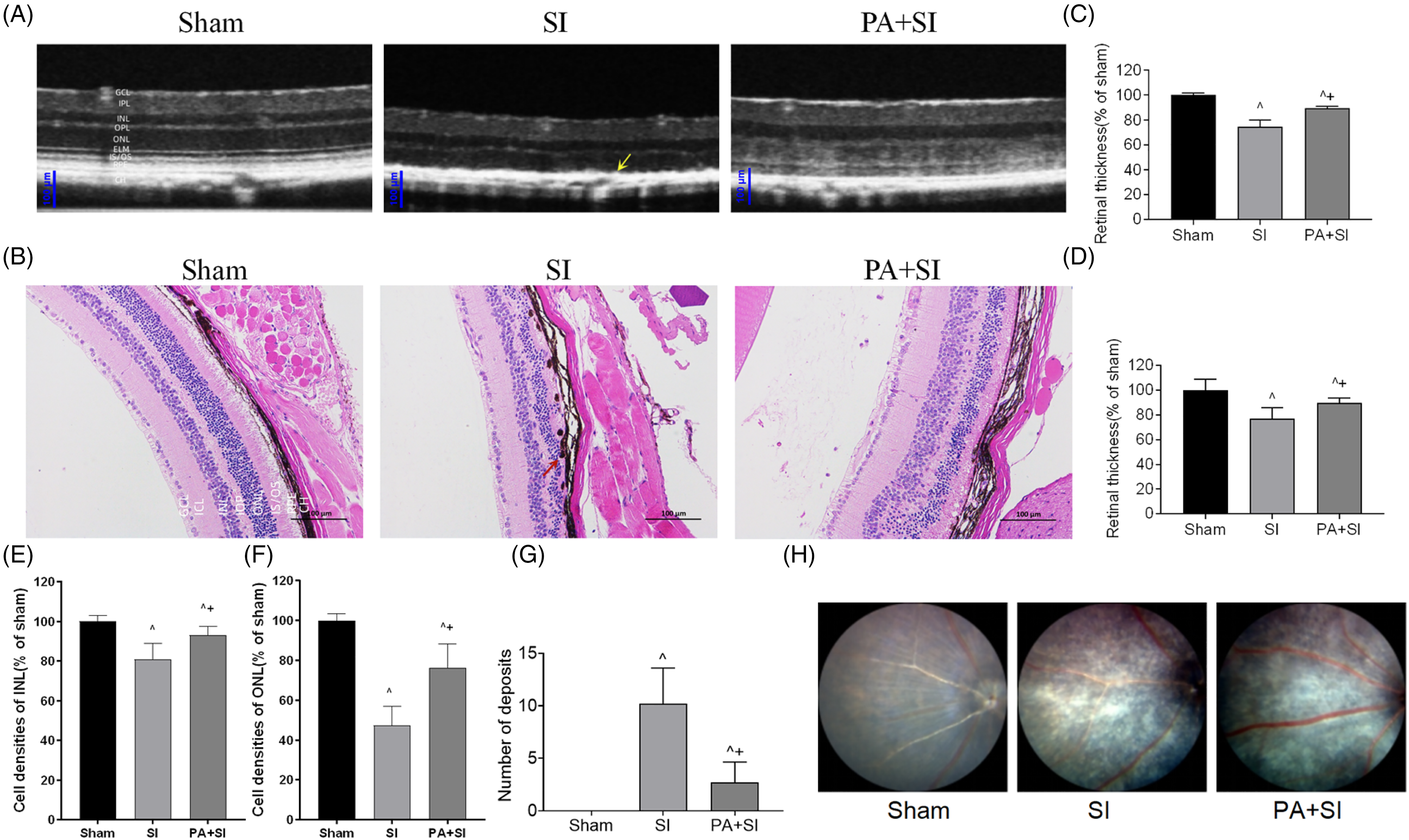
Protective effect of PA on retinal structure. (A) OCT was used to study the protective effect of PA on the retina of mice treated with sodium iodate (SI). From the OCT image, it was observed that the retina of mice in the SI group showed obvious structural abnormalities. The structure of the RPE layer and photoreceptor cell layer indicated by the yellow arrow was altered, showing a high reflection signal band with an arch back up (5 mice/10 eyes per group). (B) HE staining of the retina on day 28 after SI treatment. Sodium iodate destroys Bruch's membrane (5 mice/10 eyes per group). Scale bar: 100 μm (magnification 100 ×). (C, D) Based on the analysis from OCT and HE staining results, retinal thinning (^P < .05) was achieved in both SI and SI + PA compared to that in the sham group; however, PA significantly inhibited retinal thinning (+P < .05) at day 28 after SI treatment compared to that in the SI control group. (E, F) The cell densities of INL and ONL. (G) Both SI and SI + PA significantly increased the number of deposits compared to that in the sham group (^P <.05); however, the number of deposits was significantly inhibited after PA intervention compared to that in the SI group (+P < .05). (H) The fundus image examination (5 mice/10 eyes per group).
PA Activates the Nrf2 Pathway by Promoting the Translocation of Nrf2 into the Nucleus
The two-dimensional molecular structure of PA was investigated using the PubChem database (https://pubchem.ncbi.nlm.nih.gov/compound/5484385) (Figure 5a). The downstream target gene of PA through SwissTargetPrediction (http://www.swisstargetprediction.ch/) was used in the STRING database (https://cn.string-db.org/cgi/input) enrichment analysis. We observed that the downstream target gene of PA interacts with the Nrf2 signaling pathway-related proteins NFE2L2 and HMOX1 (Figure 5b). Signaling pathways associated with PA target genes were enriched in the Reactome database (https://reactome.org/) (Figure 5c). Furthermore, the immunofluorescence results showed that PA promoted nuclear translocation and increased the expression levels of Nrf2 and HO-1 in the RPE cells (Figure 5d and e). The immunofluorescence results showed that the intensity of Nrf2 labeled with red fluorescence was weak in both the cytoplasm and nucleus of cells in the mouse retina from the sham group. In contrast, SI treatment limited the weak red fluorescence in the cytoplasm, and PA treatment enhanced both nuclear translocation and fluorescence brightness of Nrf2 in RPE cells, as shown in Figure 5d. The protein HO-1 was marked red in parallel experiments and distributed in the cytoplasm with a blue nucleus. The SI treatment significantly reduced brightness, which was elevated by the PA treatment (Figure 5e).

PA promotes Nrf2 nuclear translocation to activate the Nrf2 signaling pathway. (A) The 2D molecular structure of PA was analyzed using PubChem. (B) Target genes of PA were analyzed using the SwissTargetPrediction database, and the association with the Nrf2 (NFE2L2)/HO-1 (HMOX1) protein was analyzed using the STRING database. (C) The signal pathways associated with PA target genes were enriched in the Reactome database. (D) Nuclear translocation of Nrf2 was detected using the immunofluorescence (IF) method (magnification 400 ×) by staining with Nrf2 antibody (red) and nuclear DAPI staining (blue) (5 mice/10 eyes per group). (E) IF method (magnification 400 ×) detection HO-1. Localization of Nrf2 and HO-1 proteins in the mouse retina after SI exposure and PA treatment (5 mice/10 eyes per group). (F-H) WB to detect Nrf2 and HO-1 protein expression after H2O2 stimulation. The experiment was repeated 3 times (*P < .05 vs control, #P < .05 vs H2O2-treated group, $P < .05 vs PA 20 μmol/L).
The expression of Nrf2 and HO-1 in H2O2-treated RPE cells increased significantly (P < .05) after 2 h of 20 μM PA pretreatment (Figure 5f to h). For further verification, the inhibitors ML385 and ZnPP were used simultaneously with H2O2 and PA, and the inhibition of Nrf2 or HO-1 blocked the anti-oxidative stress effect of PA. This suggests that the Nrf2/HO-1 pathway plays an important role in PA inhibition of RPE oxidative damage after H2O2 treatment.
Discussion
Two types of AMD were observed: dry (atrophic) and wet (neovascular). The late stage of dry AMD, which accounts for 80% to 90% of patients, is characterized by geographic atrophy (GA). 18 Currently, ongoing trials for the treatment of dry AMD include anti-complement, stem cell, and RPE replacement therapies. However, there is no treatment approved for clinical therapy. 19 Oxidative damage is the leading cause of retinal pigment epithelial degeneration in dry AMD. 20 Compounds with antioxidant properties or designed to interfere with ROS production have been proposed as potential therapeutic targets for patients with AMD. 21 In our study, we observed that PA significantly improved the activity of H2O2-treated RPE cells and inhibited intracellular ROS in vitro, which indicated that PA has a protective effect against H2O2-induced ARPE-19 cell damage. PA treatment improves cell viability and reduces the production of ROS by elevating SOD, CAT, and GSH-Px levels (key antioxidant enzymes that reflect oxidative damage in RPE cells). 22
Apoptosis is the main mechanism of death in RPE cells in response to oxidative stress and AMD.23,24 Several studies have shown that PA has anti-apoptotic effects.25,26 In our study, we observed that the apoptosis rate of ARPE-19 cells significantly increased after H2O2 stimulation and that PA treatment exerted an inhibitory effect. The expression levels of Bcl-2, Bax, and caspase-3 reflect the process of apoptosis. 27 We observed that the expression levels of Caspase-3 and Bax proteins were significantly reduced, Bcl-2 increased after PA treatment, and the MMP decreased when subjected to H2O2, which agrees with a previous study. 28 In contrast, the PA pretreatment of 20 μmol/L for 2 h significantly increased the MMP. SI-induced oxidative damage to the RPE is often used as an acute animal model to study the mechanism of oxidative damage to the RPE. 29 We used SI (40 mg/kg) to induce retinal oxidative damage in mice. Using OCT, we observed that after SI administration, the mouse RPE was disordered with deposits and thinning. PA treatment relieved retinal thinning. The histopathological observations supported the OCT results.
Elevating Nrf2 expression promotes the recovery of retinal function and protects retinal cells from oxidative damage.30,31 Therefore, targeting Nrf2 may be a potential therapeutic strategy for dry AMD. Numerous studies have confirmed that the Nrf2/HO-1 signaling pathway is pivotal in the development of AMD by protecting it from oxidative damage to RPE cells.32,33 A novel triterpenoid RTA 408 showed a protective effect against H2O2-induced cell injury via Nrf2 activation in human RPE cells. 34 Another triterpenoid Nrf2 activator, RS9, promoted autophagocytosis of the photoreceptor outer segments. 35 PA has been observed to reverse pulmonary hypertension by modulating the Nrf2-mediated signaling pathway. 36 Some studies have reported that PA regulates Nrf2/HO-1 by targeting miR-155 signaling pathway to prevent neuronal injury caused by hypoxia/reoxygenation. 37 PA showed a stronger anti-angiogenesis ability by inhibiting MMP3. 38 The activation of the PI3K/Akt signaling pathway by PA has obvious neuroprotective effects on cerebral I/R injury and neuronal apoptosis. 26 In our study, PA upregulates the expression of Nrf2 and HO-1 proteins, as shown by fluorescence staining in H2O2-induced ARPE-19 cells. We observed that ML385 and ZnPP (the inhibitors of Nrf2 and HO-1) reversed the protective effect of PA on cell death in H2O2-induced RPE cells. However, the inhibitory effects of these 2 compounds are different. PA could activate Nrf2, thereby upregulating the expression of the downstream antioxidant gene HO-1 protein, enhancing the ability of cells to clear ROS, maintaining the balance of the redox state in cells, inhibiting the oxidative stress response, and protecting cells from oxidative stress damage.
Although we discovered that PA protects against retinal injury and SI-induced retinal pigment epithelial cells, issues still need to be resolved. Although an intraperitoneal delivery of PA was used in this study, the pharmacokinetic characteristics of PA in the retina were not measured. To better understand the role of PA in dry AMD, it is necessary to investigate the molecular mechanisms of PA and its receptors, or the binding targets in AMD.
Conclusion
In conclusion, our results showed that PA protects the retina by activating the Nrf2/HO-1 pathway that inhibits the production of reactive oxygen species, increases antioxidant production in vivo, and reduces apoptosis. These results suggest that PA may play a protective role in AMD by inhibiting oxidative stress levels.
Footnotes
Authors’ Contributions
Wei wei carried out the main conception and experimental design of the study. Chengyao Qin performed most of the experiments and collected and collated the data. Xi Chen and Tianming Hu analyzed the experimental data. Yan Shao and Wenxiu Sun interpreted the experimental data. Zehao Liu and Min Li performed checks on the manuscript. All the authors read and approved the final manuscript.
Data Availability
The original contributions presented in the study are included in the article, and further inquiries can be directed to the corresponding authors.
Ethical Approval
Experimental research on vertebrates has complied with institutional guidelines and The Basel Declaration, and where available has been approved by an appropriate Ethics Committee of Liyang Traditional Chinese Medicine Hospital (No. 2023LY-1-20-01).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The present study was supported by the National Natural Science Foundation of China (No.81774370), the Changzhou Science and Technology Bureau (CJ20239002), the Liyang Science and Technology Bureau (LC2021002).
Statement of Human and Animal Rights
Experimental research on vertebrates has complied with institutional guidelines and The Basel Declaration, and where available has been approved by an appropriate Ethics Committee of Liyang Traditional Chinese Medicine Hospital (No. 2023LY-1-20-01).
Statement of Informed Consent
Experimental research on vertebrates has complied with institutional guidelines and The Basel Declaration, and where available has been approved by an appropriate Ethics Committee of Liyang Traditional Chinese Medicine Hospital (No. 2023LY-1-20-01).