
Abstract
Since late 2019 to early 2020, an outbreak caused by severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2) has become a worldwide health emergency due to its rapid infection and mortality of millions of people around the world. As the main protease Mpro or 3CLpro produced by the virus plays an important role in coronavirus survival and proliferation, it becomes an excellent drug target to identify COVID-19 inhibitors. Lawsonia inermis L. (henna) is a medicinal plant that has been used for a long time for the treatment of many fungal and bacterial infections. In the search for new anti-COVID agents from medicinal plants, we report the results of our study into the potential inhibition of Mpro by the compounds isolated from the extracts of L. inermis roots and leaves using molecular docking and molecular dynamics simulation. The molecular modeling results showed that all isolated compounds bonded spontaneously into the catalytic pockets of Mpro with binding energies <0. The docking and calculated pharmacokinetic results of the compounds (
Introduction
A novel deadly coronavirus known as severe acute respiratory syndrome-coronavirus 2 (SARS-CoV-2) has led to a worldwide health emergency. 1 The infection caused by this virus to the lungs and respiratory system is so severe that it has already killed more than 5.9 million people around the world till 24 February 2022 and infected more than 428 million people since its first case was reported in December 2019 in Wuhan city of China. 2 The virus produces many kinds of enzymes to enable it to invade the lung cells of humans. There are several druggable enzymes/proteins of SARS-CoV-2 that have attracted the attention of scientists around the world as research targets, including the nonstructural protein (nsp 1-16), RNA polymerase, helicase, the main structural proteins (spike [S], membrane [M], envelope [E], nucleocapsid phosphoprotein [N]), and papain-like protease (PLpro). 3 Out of these drug targets, the main focus has been on Mpro or 3CLpro because they act as essential multitasking polyproteins for virus survival, to facilitate the vital processes like replication, transcription, and translation of viral RNA into functional non-structural proteins.1,4 Inhibiting the activity of this Mpro protease would block viral proliferation and invasion. 5 Hence, this protease has become an excellent drug target among all the proteins of the coronavirus to identify COVID-19 inhibitors. In addition, natural products from medicinal plants have become an important source for the discovery of therapeutic agents, especially in the fight against COVID-19. 6 By using computer-aided techniques for the prediction of ligand–target interactions, 7 many natural compounds of different classes, such as baicalin, chrysin (Scutellaria baicalensis) (flavonoids), sugetriol-3,9-diacetate (Cyperus rotundus) (sesquiterpenoid), phaitanthrin D, 2,2-di (3-indolyl)-3-indolone (Isatis indigotica) (alkaloids), and platycodin D (Platycodon grandiflorus) (triterpenoid) have been reported to be potential new scaffolds in the treatment of SARS-CoV-2 as they exhibited high binding affinity to Mpro protein.6,8
Lawsonia inermis L. (syn. L. alba Lamk.), known as henna or hina, is a flowering plant belonging to the family Lythraceae that was first scientifically described by Carl von Linné (Linnaeus) in 1753. The species is named after the Scottish physician Isaac Lawson, a good friend of Linnaeus. 9
The henna plant has been widely grown in many countries and continents around the world for more than three thousand years, with a variety of uses, including ornamental, dyeing, and medicine. 10 Henna has also been used for a long time as a colorant in many countries. Henna is used as a body dye, especially in India and the Middle East, for tattoos, hair, beard, eyebrows, skin decoration, and nails. Henna powder is mixed with shampoo and other hair dyes in different proportions to produce colors ranging from bronze to reddish. It is also used for fabric dying, including silk, textiles, wool, and leather. In folk medicine, the bark is traditionally used in the treatment of jaundice, spleen enlargement, renal calculus, leprosy, and obstinate skin diseases. In Morocco, the infusion of leaves of L. inermis is used against diarrhea, renal lithiase, and gastric pains. 11 In Vietnamese traditional medicine, it is used to treat menstrual disorder, edema, rheumatism, bronchitis, and hemorrhoids. 12 Henna is used in the treatment of hair and scalp problems and for hair loss. 13 The plant has properties that effectively prevent the growth of bacteria, 14 prevent and treat fungal infections, have analgesic and anti-inflammatory effects on the human skin, and are effective against lice and sunburn. 10 ,15 Phytochemical studies of leaves, barks, and flowers of L. inermis revealed the presence of many kinds of compounds including flavonoids, alkaloids, terpenoids, sterols, and especially triterpenoids.10,16
In the search for new inhibitory scaffolds from Vietnamese medicinal plants for the treatment of the SARS-CoV-2 triggered global health coronavirus disease 2019, this study was designed to evaluate the potential biological activity of natural compounds (
Materials and Methods
General Experimental Procedures
1H-NMR (500 MHz) and 13C-NMR (125 MHz) spectra were measured on a Bruker AVANCE 500 MHz spectrometer with tetramethylsilane (TMS) as an internal standard, and chemical shifts are expressed in ppm. ESI-MS were obtained from a Varian FT-MS spectrometer and MicroQ-TOF III (Bruker Daltonics Inc.). Optical rotations were measured on DIP-2000 KUY polarimeter (JASCO, Tokyo, Japan), and UV and IR spectra on a UV-VIS spectrophotometer (Varian Cary, Australia) and a FT-IR spectrometer 1650-Perkin Elmer (USA), respectively. Column chromatography (CC) was carried out on silica gel (Si 60 F254, 230-400 mesh, Merck). All solvents were redistilled before use. Analytical and preparative TLC were performed on pre-coated Kieselgel (Si 60 F254) or RP18 plates (0.25 mm, Merck). Compounds were visualized under UV radiation (254, 365 nm) and by spraying plates with 10% H2SO4, followed by heating with a heat gun.
Plant Materials
L. inermis whole plants (roots, leaves, and stems) were collected from in Ngoc Truc Village, Dai Mo Commune, Ha Dong District, Hanoi. Specimens was identified at the National Institute of Medicinal Materials (NIMM), and a voucher specimen (HNIP/16698/08) was deposited at the Herbarium of the Department of Botany at Hanoi University of Pharmacy (HUP), Hanoi, Vietnam.
Extraction and Isolation
The roots of L. inermis were dried in the shade at room temperature and ground into powder. Dried powdered roots (3.0 kg) were extracted with MeOH (3 × 5 L) over 5 days at room temperature. The methanol extracts were collected and concentrated under reduced pressure to yield a black crude MeOH extract (20 g). This was suspended in hot water (1:1, v/v) and successively partitioned with n-hexane (H), chloroform (C), ethyl acetate (E), and n-butanol (B). The resulting fractions were concentrated under reduced pressure to give the corresponding n-hexane (2.1 g), chloroform (2.9 g), EtOAc (8.8 g), and n-butanol (3.2 g) fractions, which were kept at 4 °C in the dark until further analysis.
The chloroform fraction (2.8 g) was subjected to normal phase silica gel CC eluting with n-hexane/CH2Cl2/MeOH (gradient mixtures 7:3:0.5-3:7:0.5) to afford three fractions (CR1-3). Fraction CR1 was repeatedly chromatographed over a silica gel column and eluted with n-hexane/CH2Cl2 (8:2 v/v) to obtain compounds
The same extraction procedure was applied to the dried leaves of L. inermis. Briefly, the leaf powder (5.0 kg) was extracted with EtOH 85% (3 × 5-10 L) (3 × 24h). The ethanol extracts were collected and concentrated under reduced pressure to yield a black crude extract (31 g), which was then suspended in hot water (1:1, v/v) and successively partitioned with n-hexane (H), chloroform (C), ethyl acetate (E), and n-butanol (B). The resulting fractions were concentrated under reduced pressure to give the corresponding n-hexane (1.4 g), chloroform (7.0 g), EtOAc (5.4 g), and n-butanol (9.2 g) fractions.
The chloroform fraction (7.0 g) was chromatographed over a silica gel column eluted with gradient mixtures (CHCl3/MeOH [0%-100%-50/50%, 2L/each gradient]) to obtain 4 sub-fractions (CL1-4). Fraction CL4 was placed in a fridge at −4 °C overnight. The yellow precipitate that formed was re-crystallized in cold EtOAc and re-filtered to afford compound
The n-hexane fraction H (2.1 g) was chromatographed over silica-gel CC, eluting with n-hexane/EtOAc (6:4-2:6) to obtain compounds
Molecular Docking Analysis
Molecular docking was performed with AutoDock v4.2 and AutodockVina v1.2.3.16,17 The computational software was downloaded from the website http://scripps.edu, operated under Microsoft Windows 8, installed on an Intel i7 PC with a 3.2 GHz processor and 16 GB RAM.
Phytochemicals Optimization
Ligand structures (
Protein Preparation
The main protease of SARS-CoV-2 in the form of three-dimensional crystal structures bound to a co-crystal ligand (Mpro + O6K, in orthorhombic form) were downloaded from the Protein Data Bank (www.rcsb.org) (PDB ID: 6y2g) with a resolution of 2.20 Å.
5
All crystallized water molecules, heteroatoms, and the bound inhibitor present in the protease crystal structure were removed by BIOVIA Discovery Studio Visualizer 2021
18
and AutoDockTools.
16
The torsions were fixed for the ligand. The rigid grid box was attained using Autogrid. A grid box (21.1 Å × 21.4 Å × 24.3 Å) with coordinates x = −19.9, y = −6.2, z = − 26.8 and exhaustiveness = 400 was set around the active site, where the protein acid amines interacted with its co-crystal
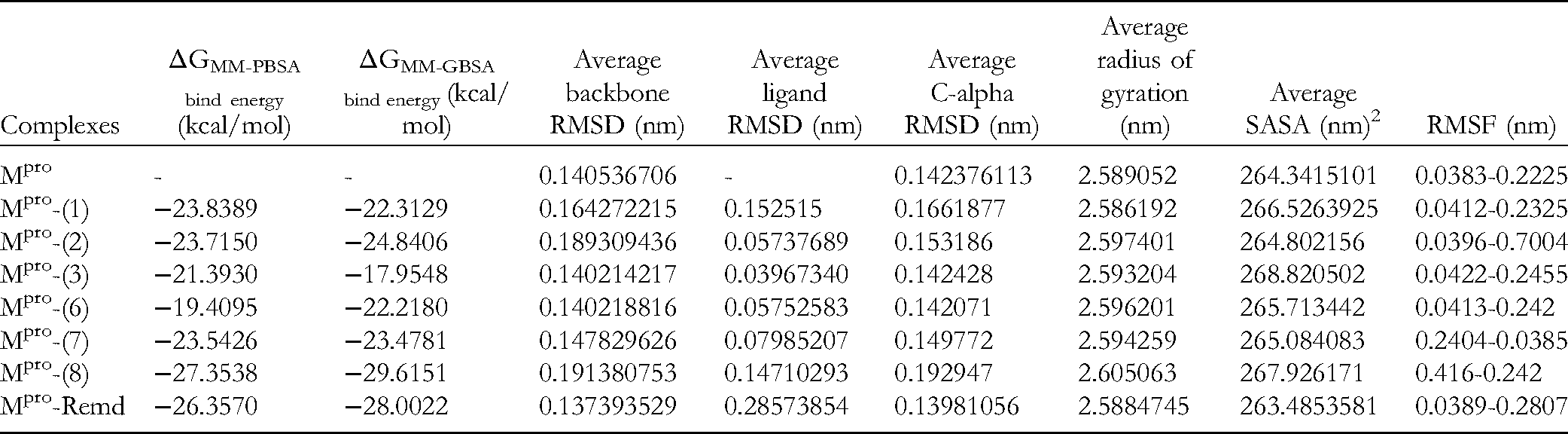
Calculation of Binding Free Energy Using MM-PBSA and MM-GBSA and Average Values of RMSD (Backbone, Ligands, and C-Alpha), Rg (Radius of Gyration), SASA, and RMSF of Mpro Complexes with Tested Compounds (
Molecular Dynamics Simulation
From the results of molecular docking simulations, we conducted molecular dynamics (MD) simulation of the selected compounds with the potential to inhibit SARS-CoV-2 Mpro, including
The free binding energy calculation in MD was performed by the gmx_MM-PBSA protocol based on the single trajectory of GROMACS with AMBER force field. 34 The binding free energy ΔGbind of each complex in water was estimated via the commonly used Molecular Mechanics-Poisson − Boltzmann Surface Area (MM-PBSA) and Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) methods. 35 A total of 5000 snapshots were the output from the trajectory every 10 ps. The free binding affinities and the RMSD were analyzed. Other parameters, like RMSD score (backbone, ligand, C-α), root-mean-square fluctuations (RMSF), radius of gyration (Rg), and solvent accessible surface area (SASA) calculations, were carried out with GROMACS tools.
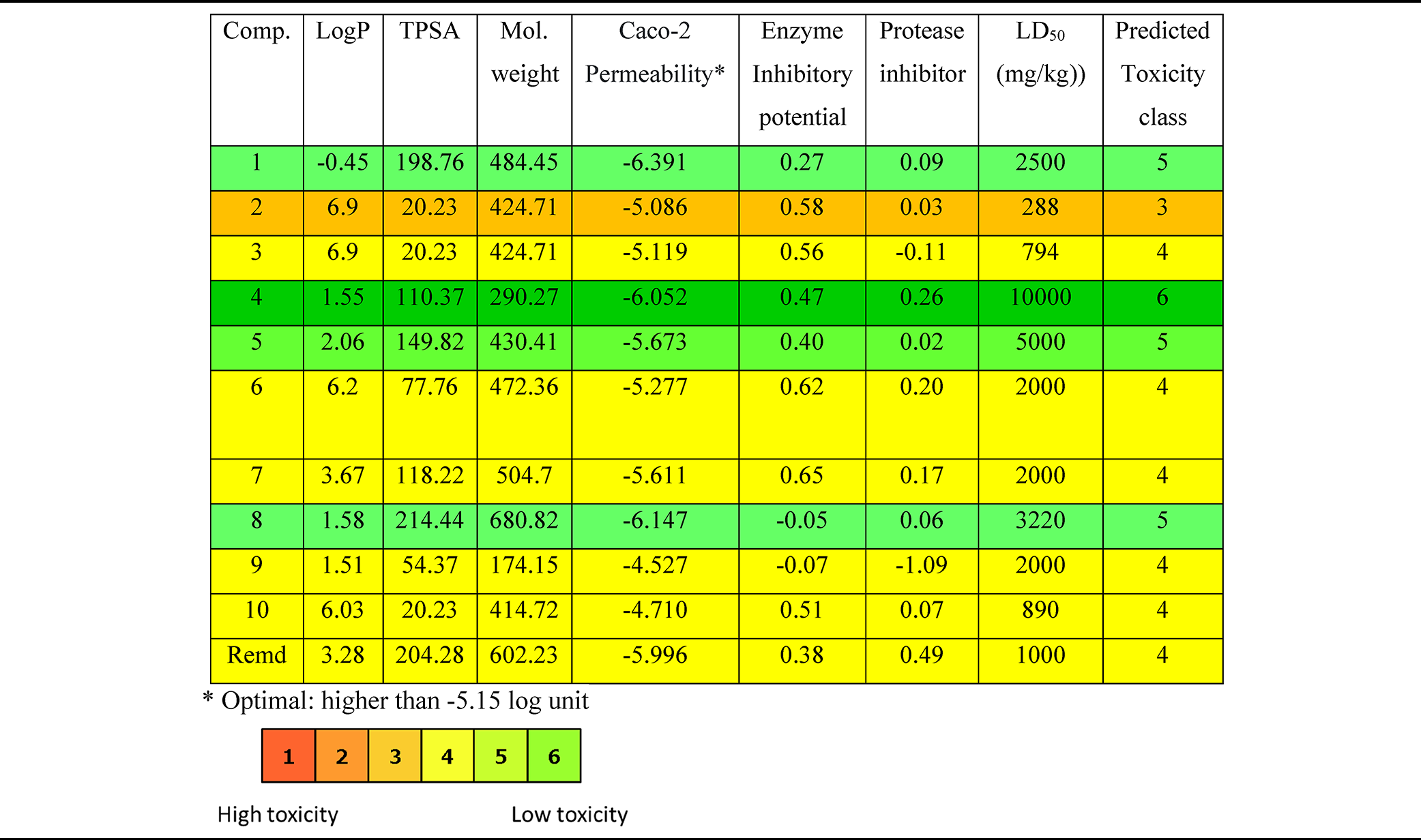
ADMET Analysis
The toxicity profiles were verified for each candidate using ProTox (http://tox.charite.de/protox_II/) where data like hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, toxicity class, and possible lethal dose (LD50) value were calculated. 36 Furthermore, the overall drug-likeness characteristics were performed by in silico ADMET study (absorption, distribution, metabolism, excretion, and toxicity) of each compound measured using tools such as Molsoft (https://molsoft.com/mprop/), Admetlab 2.0 (https://admetmesh.scbdd.com/), SwissADME (https://www.swissadme.ch/), and Molinspiration (https://molinspiration.com/) to identify safe and effective drug candidates in the early stage of drug discovery, pharmacokinetic study, and data analysis.
Results and Discussion
Chemical Compositions of L. inermis
From the methanolic extract of L. inermis (roots, leaves, and stems), after fractionation with solvents of different polarities: n-hexane, chloroform, ethyl acetate, and n-butanol, and repeated chromatography on silica gel, dianion HP-20 and reversed phase RP-18, 12 known compounds were isolated (

The structures of isolated compounds
Molecular Docking Analysis
Compounds
Identification of Active Binding Site
As a first approach to identify the best poses within the binding pocket of Mpro

(A) Cartoon representation of 3 proposed active binding sites (green, pink, and blue lines) of Mpro protease 6y2g by FTSite server. (B) The result of re-docking ligand O6K (co-crystal) into the active site region of Mpro to validate docking parameters. The redock ligand (blue layer) overlapped the original ligand (native) (green). (C) The superimposition of isolated compounds (
Next, in order to validate the docking parameters and to examine the active binding site, we investigated the crystallographic structures of Mpro complexed with inhibitors and redocked the co-crystal
Binding Parameters
After strengthening the binding site, the 10 compounds were docked to the Mpro protein and the docking simulation results highlighted that the isolated compounds (

Network of binding interactions of compounds
Free Binding Energy (ΔG, kcal/mol) and IC50,calcd. (μM [Micromolar]) and Network of Binding Interactions Calculated by Molecular Docking.

At temperature = 298.15 K.
The result of the molecular docking analysis indicated that the free binding energies of the compounds to Mpro ranged between − −8.19 and −5.49 kcal/mol (Table 1). The negative values of ΔG indicate that the compounds spontaneously bind to Mpro and, therefore, could have potential inhibitory activities on the Mpro enzyme (Figure 4). The molecules occupied the same cavity, with only some differences in the involvement of amino acid residues. The variance of the ΔG values and the placement of the bond inside the active pocket could be caused by the functional groups presented in the compounds. In Table 1, the docking results of the isolated compounds (ligands) from L. inermis, especially of the five triterpenoids, with the main protease (Mpro) of SARS-CoV-2 were remarkable; the calculated binding energy of

Hydrophobic interaction and hydrogen bonds between Mpro and the selected triterpenoids (
Correspondingly, the inhibition concentrations induced by these triterpenoids were with IC50,calcd. values of 0.98, 2.13, 3.96, 4.64, and 6.44 μM, respectively. In particular, compound

2D and 3D presentations of interactions between compound
Pharmacokinetic Parameters and Toxicity Prediction of the Isolated Compounds (

Network Binding Interactions
Additionally, a network of binding interactions of the tested compounds (
MD Simulation
The binding energy was less than 0 kcal/mol, indicating that the ligand and receptor can bind spontaneously and the complex formed between the active compounds and the target might have greater stability. Therefore, based on the results of molecular docking analysis, the compounds
After MD simulation for a period of 50 ns (Figure 6), the complexes were refined and their overall energy and RMSD score, RMSD of backbone, RMSD ligands, RMSF, RMSD C-α, Rg, SASA, and H-bond were obtained (Figure 6A to H and Table 2).

Time plot of (A) RMSD backbone, (B) RMSD ligand, (C) 2D interaction of compound 8 with Mpro protein at the end of the simulation time of 50 ns, (D) RMSF, (E) RMSD C-alpha, (F) Rg, (G) SASA, and (H) H-bond of 5 Mpro–inhibitor complexes.
The overall binding free energies (ΔG bind energy) of the Mpro-inhibitor complex systems were calculated using the MM-PBSA and MM-GBSA methods from 500 snapshots and from the whole MD trajectories, in which ligand and protein can assume different conformations.
35
The estimated values of binding energy ΔGMM-PBSA calculated for Mpro-(
The RMSD backbone is the measure of the average distance between the atoms of the tested compounds to the Mpro protein backbone. The average RMSD backbone values of Mpro-(
During the simulation time of 50 ns, the average RMSD ligand value of the Mpro-inhibitors systems was measured to be in the average range of 0.039-0.147 nm (Table 2). RMSDs of ligands in complexes Mpro-(
As the value of RMSF represents the flexibility of conformational residues, the fluctuation in the RMSD backbone and ligand of the Mpro-(
The RMSD trajectories of the C-α atom were described for the dynamics and convergence of each Mpro-inhibitor system (Figure 6E). It was shown that the C-α RMSD values of Mpro-(
Additionally, in order to evaluate the compactness of the Mpro-inhibitor system after MD simulation, we further calculated the radius of gyration (Rg). Figure 6F showed that the Rg of Mpro is observed to be nearly stable in the consistency of the oscillations throughout the simulation. The Rg value of the complex Mpro-(
The interactions between the Mpro-inhibitor complex system and the aqueous solvent were studied by SASA during the time of MD simulation. This was aimed to investigate the degree of structural change that occurred after the interaction. Figure 6G shows the plot of the SASA value versus time for all Mpro-inhibitor complexes. The mean SASA values gradually decreased from 264.802156 nm2 (Mpro-
The number of intermolecular hydrogen bonds in the ligand-protein complex was determined, which contributes to the stability of the structure. It is shown in Figure 6H that hydrogen bonds are present in all Mpro-inhibitor complexes. The highest number of hydrogen bonds was observed for the Mpro-
ADMET Analysis
Additionally, pre-clinical toxicity studies are important to establish the margin of safety and to consider efficiently the risk-benefit of a proposed drug
7
so that pharmacophoric features influencing the behavior of a molecule in a living organism need to be calculated, including bioavailability (LogP—octanol/water partition coefficient), transport properties (total molecular polar surface area [TPSA]), Caco-2 permeability, enzyme and protease inhibitor potential, toxicity predicted LD50 (mg/kg), and predictive toxicity class of the compounds (Table 3). Generally, the tested compounds were predicted to be in toxicity class from 3 (medium-toxic) to 6 (non-toxic). Compound (
Conclusion
From the different parts of L. inermis, 12 compounds were isolated, including 5 triterpenoids (
Supplemental Material
sj-docx-1-npx-10.1177_1934578X221125161 - Supplemental material for Compounds Isolated from Lawsonia inermis L. Collected in Vietnam and Evaluation of Their Potential Activity Against the Main Protease of SARS-CoV-2 Using In silico Molecular Docking and Molecular Dynamic Simulation
Supplemental material, sj-docx-1-npx-10.1177_1934578X221125161 for Compounds Isolated from Lawsonia inermis L. Collected in Vietnam and Evaluation of Their Potential Activity Against the Main Protease of SARS-CoV-2 Using In silico Molecular Docking and Molecular Dynamic Simulation by Nguyen Anh Tuan, Pham Ngoc Khanh, Nguyen Xuan Ha, Ta Chi Binh, Nguyen Duy Khanh and Tran Thị Oanh in Natural Product Communications
Footnotes
Acknowledgements
This work was financial supported by the Ministry of Science and Technology for the Project “Study on production of antifungal drug for treatment of trichophyton, epidermophyton, dermatophytes from Lawsonia inermis L. in Vietnam.” The authors would like to give special thanks to Prof. Dr Nguyen Manh Cuong, INPC, who helped us to finish this paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Ministry of Science and Technology (grant number: MOST/19-22).
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.