
Abstract
Acetylcholinesterase (AChE) plays an essential role in the cholinergic pathways in Alzheimer's disease. This study used a deep learning model as a powerful virtual tool for discovering AChE inhibitors. The model showed 94.3% accuracy, 97.1% precision, 95.9% recall, and 86.2% specificity. A list of bioactive compounds extracted from Pongamia pinnata (L.) Pierre was selected as the test dataset. Four candidates were selected for in vitro: pongapin, ovalichromene B, gamatin, and pongaglabrone. These flavonoids showed inhibitory effects with half-maximal inhibitory concentration (IC50) values between 19.8 and 63.5 μg/mL. In molecular analyses, these compounds showed noticeable interactions with the AChE catalytic residues Ser203 and His447 and satisfied acceptable drug-like properties and other druglikeness parameters. This study has shown that a deep learning approach can accurately predict potential compounds targeting AChE, and P. pinnata is a promising medical plant for Alzheimer's disease.
Introduction
Alzheimer's disease is a common cause of dementia, and the disease is being described for the first time in the early 20th century by the psychiatrist Alois Alzheimer. Patients progressively lose their memory and ability to focus. In addition, their ability to navigate becomes increasingly complex, making it difficult to perform daily activities. However, the impact of Alzheimer's disease on patients can be reduced by different drugs and non-pharmacological methods. 1 Therefore, developing natural compounds that influence the therapeutic process in Alzheimer's patients has been identified as a potential future research direction.
Acetylcholine (ACh), found in the neural system of many animals and humans, acts as a neurotransmitter. Acetylcholinesterase (AChE) is a cholinergic enzyme that catalyzes the hydrolysis of ACh neurotransmitters into ACh or other choline esters. Several studies have investigated AChE in the tissues related to Alzheimer's disease, showing changes in AChE activity in the brain, cerebrospinal fluid (CSF), and blood of Alzheimer's patients.2,3 AChE and other enzyme components form a plaque-causing complex in older adults. In addition, the neurotoxicity of amyloid is increased in the presence of AChE. 4 Therefore, the search for AChE inhibiting compounds has been identified as a potential research direction.
Drug discovery and development is a complex and expensive process. It costs over US$2.6 billion to develop a new drug. 5 Computational methods have been used to discover new pharmacological candidates through virtual screening analyses.6‐8 Recent advances in computer science may accelerate and improve pharmaceutical research and development. Deep learning is a form of artificial intelligence (AI) that uses the power of computers and algorithms to solve tasks expediently. 9 Rapid and remarkable progress has been made on various medical problems through deep learning and large-scale annotated datasets.10‐12
Medicinal chemistry has adopted various forms of deep learning in drug design with varying degrees of success. For example, many drug features are predicted using the quantitative structure–activity relationship (QSAR) method, such as solubility and bioactivity. Recently, many public datasets have been made available for screening and testing the activity of chemical compounds in drug development and research and are particularly useful for training models to predict the bioavailability of other compounds. The implementation of deep neural networks has resulted in an unprecedented acceleration in this field. This study uses a deep learning approach to classify chemical compounds with bioactivities on AChE.
Pongamia pinnata belongs to the Fabaceae family. Previous studies have found its oil to possess activities useful for treating tumors, skin diseases, abscesses, arthritis, wounds, ulcers, diarrhea, hyperglycemia, and blood lipids.13‐15 In addition, the toxicity and inhibition of P. pinnata for pests such as Pediculus humanus capitis, Callosobruchus chinensis, and Odototermes obesus have been studied in the laboratory.16‐19 Importantly, compounds isolated from P. pinnata have shown anti-Alzheimer's activity. 20 However, the biological activity of P. pinnata on AChE remains unknown. Therefore, this study uses the deep learning model and AChE inhibition assay to screen potential natural AChE inhibitors isolated from P. pinnata.
Results and Discussion
Deep Learning Model
A dataset of published experimental experiments was retrieved from the ChEMBL database and previous studies.21,22 Half-maximal inhibitory concentrations (IC50) were converted to μg/mL. The dataset was classified into 2 groups using a cut-off value of 50 μg/mL: active (IC50 < 50 μg/mL) and inactive (IC50 ≥ 50 μg/mL). The division of the dataset into the 2 groups was unbalanced. Therefore, we used resampling for the training dataset to account for the unbalanced groups. We developed a 1D convolutional neural network (CNN) model to predict 2 classes of bioactive compounds inhibiting AChE using the compound's molecular descriptors. The structure of the proposed deep CNNs is presented in Figure 1. The proposed CNN model consists of 6 main convolutional layers, following 6 activation layers and 6 pooling layers. The previous layers are used for feature extraction, after which we added neural and dropout layers (0.3). The activation layers are connected by a rectified linear unit (ReLU) function. Finally, the last layer has 1 node connected to the previous layer by the softmax activation function.

The structure of the deep convolutional neural model.
The model was trained using 1000 epochs with batch_size = 25. The results showed that the accuracy of the training and validation datasets was above 90% (Figure 2).

Accuracy and loss of deep learning model (1000 epochs).
It can be seen that after the model was trained for 200 epochs, the accuracy rate remained unchanged. However, while the training loss has remained virtually unchanged, the validation loss increases. Therefore, the model may be overfitting when trained using many epochs. Therefore, we decided to train the model with 200 epochs to optimize its parameters.
The model was trained using 200 epochs with batch_size = 25, and learning_rate = 0.0001. The results showed that the accuracy of the validation dataset is 94.3%, and the validation loss is similar to the training loss (Figure 3). The confusion matrix of the validation dataset is presented in Table 1.
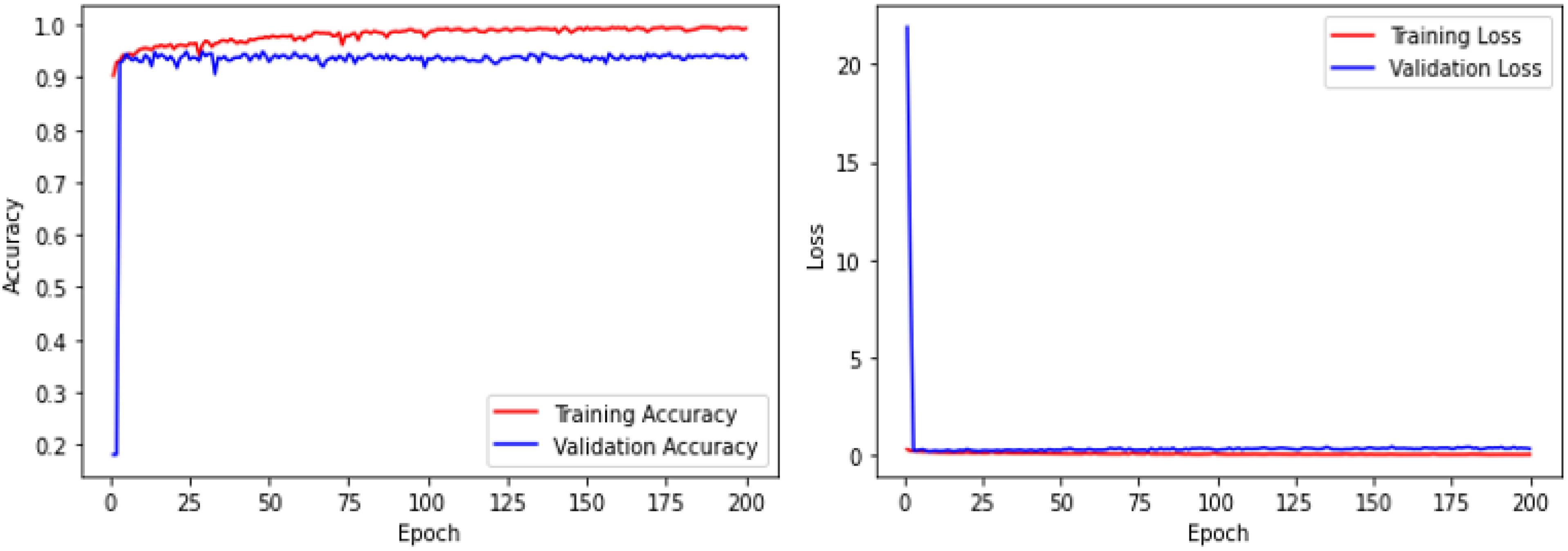
Accuracy and loss of deep learning model (200 epochs).
The Confusion Matrix of the Validation Dataset.

Following the confusion matrix, we computed the average values of accuracy, precision, recall, and specificity, defined as follows:
The average results showed 94.3% accuracy, 97.1% precision, 95.9% recall, and 86.2% specificity (Table 1), highlighting the model's predictive capability. Furthermore, the screening method identified candidates with effective biological activity that can reduce the time and cost of unsuccessful testing in the experimental research phases. This result indicates that our model favors active compound selection over inactive compound selection.
In this study, our model showed precision and recall values greater than its accuracy values, indicating that the deep learning model can be used for the virtual screening of compounds that inhibit AChE. Therefore, this deep learning model was used to screen natural compounds isolated from P. pinnata; 4 compounds were identified and categorized in the “active” group, which were selected for the in vitro experiments: pongapin

Chemical structure of the 4 compounds.
AChE Assay
The AChE inhibitory activities of the 4 selected compounds were assessed at various concentrations using galanthamine as the positive control. All 4 compounds showed inhibitory activities against AChE with IC50 < 100 μg/mL. Among them, pongaglabrone (
Molecular Docking Analysis
Molecular docking is a critical method for determining the interaction between proteins and drug candidates.6‐8 We used the AutoDock Vina software to perform molecular docking to determine the structural and activity interactions between AChE and the candidate compounds. The 3D crystal structure of AChE (protein data bank [PDB] ID: 4EY7) was selected as the representative sample for further structure–activity relationship study. The active site of human AChE is located at the bottom of a 20 Å deep, narrow gorge found in different distinct domains.23,24 The peripheral anionic site is located near (18 Å) the active gorge's site. In this study, the grid box was set to the protein's active site based on the interaction site of Donepezil, the crystal ligand of the protein crystal (PDB ID: 4EY7). First, to demonstrate that the selected protein structure (PDB ID: 4EY7) and docking protocol are reliable, the ligand was re-docked to the target protein in the crystal structure, and the root-mean-square deviation (RMSD) of docking pose against the crystal pose was assessed. The result showed that the docking pose has a similar conformation and orientation as the crystal pose with an RMSD of 0.316 Å (Figure 5). Previous studies have considered an RMSD within 2.0 Å to be suitable and valuable for docking experiments, indicating that this docking protocol is suitable for use in our study.

Docking (yellow) and crystal (green) poses of the ligand in the crystal structure of acetylcholinesterase (AChE).
The molecular docking method was used to analyze the mechanical interaction of AChE with compounds

Interactions between ovalichromene B and acetylcholinesterase (AChE).
Compounds
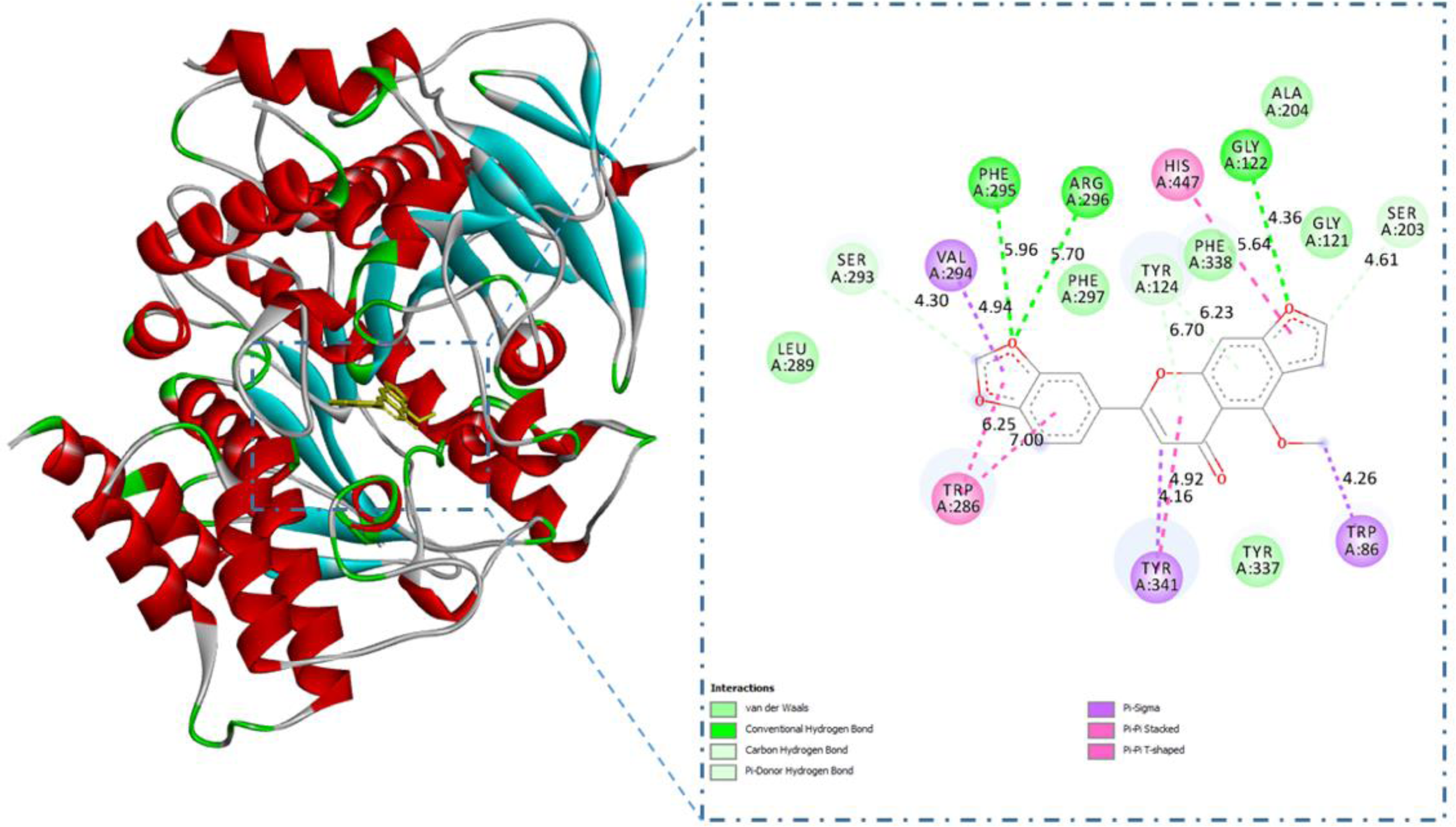
Interactions between gamatin and acetylcholinesterase (AChE).

Interactions between pongaglabrone and acetylcholinesterase (AChE).
These interactions are similar to those between pongapin (

Interactions of pongapin and acetylcholinesterase (AChE).
The interaction analysis showed that the 4 compounds (
Docking Results of 4 Flavonoids Toward Acetylcholinesterase (AChE) Protein.

Absorption, Distribution, Metabolism, and Excretion Properties
Lipinski's rule of five is a method for determining the druglikeness of active chemicals and determining whether they are orally active or not. The oral bioavailability of drugs with good membrane permeability and hydrophobicity is shown by the Log P, TPSA, MW, HBA, and HBD values, with appropriate nRotB and MR values indicating that medicines are absorbed well in the intestine and have a high oral bioavailability.
The results presented in Table 3 show that the 4 compounds possess acceptable drug-like properties and other druglikeness parameters (MW ≤500 Da, Log P <5, TPSA <140 Å, nHBD ≤5, and nHBA ≤10). These results show that these compounds have the potential to become drugs with good oral bioavailability. In addition, the Log S values are between −4.61 and −4.43, indicating moderate solubility in water, and TPSA values are below 140. 25 Consequently, the 4 compounds were predicted to have excellent gastrointestinal absorption. The drug molecule's stereo-specificity is a characteristic of nRotB, which was determined to be under 10.
Drug Properties of Isolated Compounds Analyzed With SwissADME.

Abbreviations: Log P: Log of octanol/water partition coefficient; Log S, log of solubility; MW, molecular weight; nHBA, number of hydrogen bond acceptor(s); nHBD, number of hydrogen bond donor(s); MR, molar refractivity; nRotB, number of rotatable bonds; TPSA, total polar surface area.
The in silico predictions of absorption, distribution, metabolism, and excretion (ADME) are presented in Table 4. The results indicated that all compounds are predicted to show high gastrointestinal absorption and be able to permeate the blood–brain barrier, an important property for compounds acting on the central nervous system to treat Alzheimer's disease. Moreover, various cytochromes (CYPs) vital for drug biotransformation, including CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4, are known to affect drug metabolism. 26 Ovalichromene B inhibited all cytochromes and P-glycoprotein (P-gp) substrate, while gamatin, pongaglabrone, and pongapin inhibited most cytochromes, but not P-gp substrate. These results show that all 4 compounds are promising potential drugs targeting AChE to treat Alzheimer's patients.
Acetylcholinesterase (ADME) Predictions of Isolated Compounds Computed by SwissADME.

Abbreviations: BBB Per: blood–brain barrier permeability; CYP, cytochrome-P; GI Abs: gastrointestinal absorption; log Kp, log of skin permeability; P-gp, P-glycoprotein.
Conclusion
We constructed a deep learning model based on a dataset of public compounds. The model showed 94.3% accuracy, 97.1% precision, 95.9% recall, and 86.1% specificity. This model could be applicable for identifying chemical compounds that act as AChE inhibitors. The accuracy of our model was evaluated with a list of natural compounds extracted from P. pinnata. Four flavonoid compounds (
Experimental
Deep Learning Model
Data Preparation
A drug's IC50 of AChE is widely used to measure its potency. In this study, the dataset was collected from the ChEMBL database and previous studies. Anonymous and null data were dropped. The chemical formula was saved in simplified molecular-input line-entry system (SMILES) format. Molecular descriptors of a drug included 881 features (molecular fingerprints). 881 Molecular descriptors were calculated from the SMILES formula by PaDeL-Discriptor. IC50 values of AChE were categorized into “active” and “inactive” with a cut-off value of 50 μg/mL. Finally, the resultant data amounted to 5230 input vectors. Then, the dataset was divided into 80% and 20% as the training set and validation set, respectively (Figure 10).

Data description and classes.
The list of compounds extracted from P. pinnata was collected from our previous studies27,28 and these molecular features were also calculated by PaDeL-Discriptor. This dataset was used for the testing model.
Model Building
For prediction, input data consisted of chemical properties of compounds, and we split the dataset into training and validation sets. The CNN method was applied for prediction. Conv1D layers and max-pooling layers were used to import and reduce features of the dataset, then the input data were added to the neural network. The parameters are formed and improved through the training process in the training set and evaluated by the validation set. The model to predict AChE inhibitors was built by python language on Google Colaboratory.
AChE Inhibition Assay
The AChE activity was assayed by adapting a reported spectrophotometric method reported. 29 The assay was made in triplicate in 96-well microplates, and an ELISA microplate reader (Biotek, USA) was used to measure the absorbance. To assess the inhibitory impact of the samples on AChE activity, acetylthiocholine iodide (ACTI) was used as the substrate. The reaction mixture, containing 140 μL phosphate buffer (pH 8.0), 20 μL of AChE solution (0.25 IU/mL), and 20 μL of tested sample was incubated for 15 min at 25 °C. After that, 10 μL of 2.5 mM ACTI, and 10 μL of 2.5 mM Ellman's reagent (DTNB) was added to initiate the reaction. The mixture was incubated further at 25 °C for 15 min, and the absorbance at 405 nm was measured. Galanthamine was used as a positive control. All tested samples and the positive control were dissolved in 10% DMSO. The tested sample was measured at doses of 100, 20, 4, and 0.8 μg/mL and IC50 values were calculated by the program TableCurve, Version 4.0.
Molecular Docking Simulation
Protein and Ligand Preparation
The crystal structure of AChE (PDB ID: 3LII) was retrieved from Protein Data Bank (RCSB PDB). Next, water molecules were removed and polar Kollman charges were added to the protein using AutoDock tools version 1.5.6. Finally, the macromolecule was exported into a dockable pdbqt format for molecular docking.
The information on compounds of P. pinnata was collected from previous studies and 3D structures were prepared by MarvinSketch (ChemAxon, USA)
The 3D structures of the compounds
Molecular Docking
The molecular docking process on AChE was carried out using AutoDock Vina. The grid box was identified with parameters: center_x = −11.2, center_y = −42.8, center_z = −27.3 and size_x = 22.50, size_y = 18.75, size_z = 20.25. Docking scores are reported in kcal/mol. Finally, Discovery Studio Visualizer was used to visualize the molecular interactions between proteins and ligands.
In Silico Druglikeness
Prediction of druglikeness of compounds was based on the already established concept of Lipinski's rule. 30 SwissADME server was used to calculate the ADME properties of ligands which provided information about the ADME properties of the ligands. 31
Footnotes
Availability of Data and Materials
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Ethical Approval is not applicable for this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the University of Medicine and Pharmacy, Hue University (grant number 23/21).
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.