
Abstract
Malignant tumors have become a significant public health problem that severely threatens human health. Drug-targeting therapy is essential for tumor therapy, along with surgery and radiotherapy. Of the 378 novel drugs approved over the past five years, those for oncological therapy remains at the top (25%). These drugs are used to treat patients with various cancers by acting on corresponding targets, such as EGFR, JAK, BTK, IDH, and FLT3. This review examines anti-tumor agents approved between 2016 and 2020, classifying them according to indication (such as lung cancer, leukemia, breast cancer, and myeloma). These drugs are reviewed according to their route of administration, first-in-class designation, approval dates, and expedited review categories. Furthermore, this paper summarizes the targets and modes of action of the approved anti-tumor drugs while systematically discussing their synthetic routes for medicinal chemistry or industrial use, which will benefit next-generation drug discovery.
Introduction
From 2016 to 2020, 228 drugs, including 168 chemical entities, were developed by pharmaceutical companies and approved to treat various diseases. 1 These drugs include anti-infective drugs, central nervous system drugs, cardiomyopathic drugs, metabolic drugs, dermatologic drugs, gastrointestinal drugs, and oncological drugs. A record number of drugs were approved in 2018 (59), while the fewest approvals occurred in 2016 (22). However, of the different therapeutic categories, anti-cancer drugs have consistently ranked first. The past five years have witnessed the approval of 61 drugs or drug combinations, including 42 small-molecule drugs for treating various types of cancers (Figure 1).
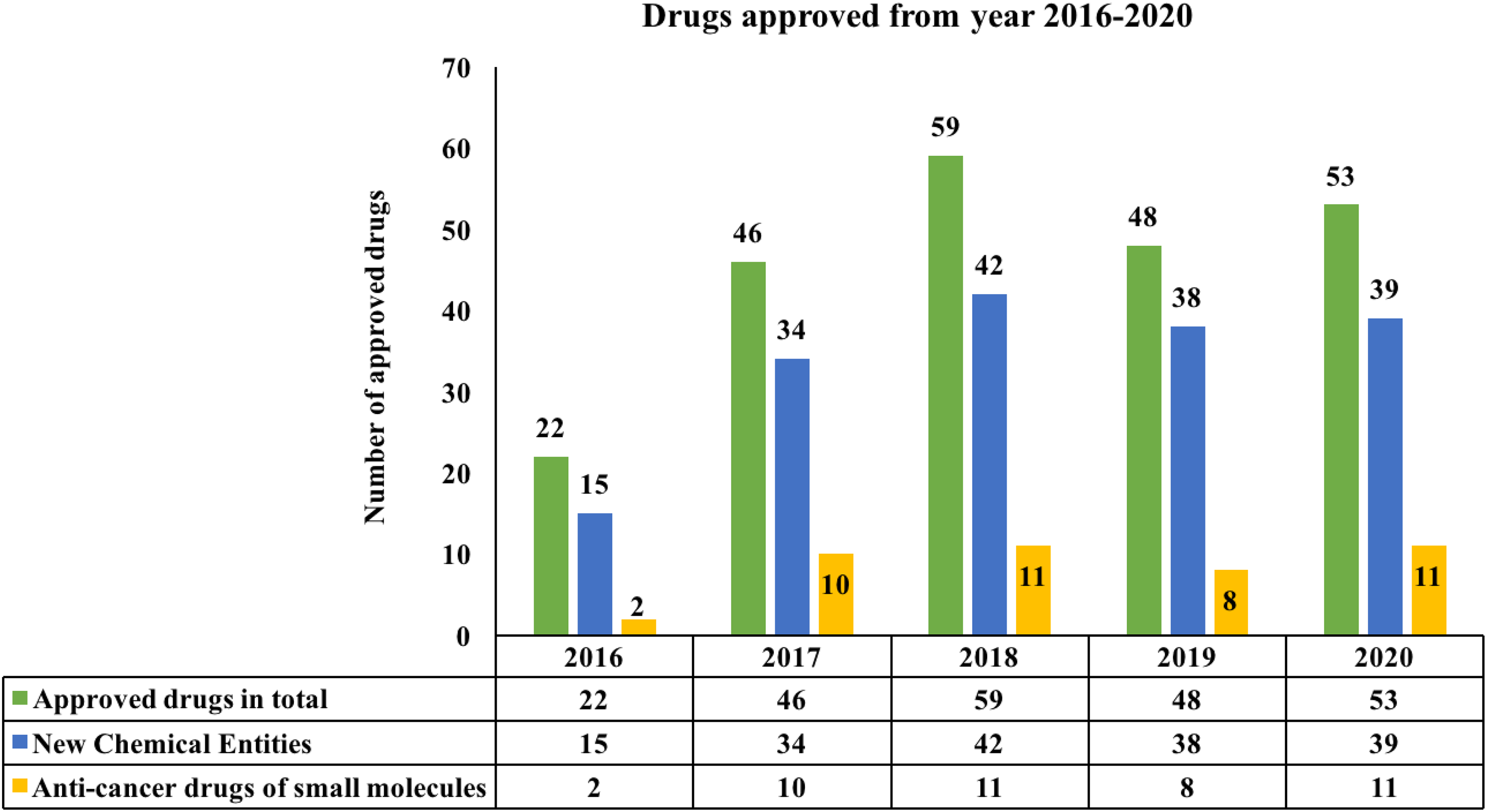
Bar graph illustration of the number of approved drugs from 2016 to 2020.
These drugs are classified according to the type of cancer, such as leukemia, breast cancer, lung cancer, prostate cancer, melanoma, gastrointestinal stromal tumors (GIST), lymphoma, myeloma, and others (Figure 2). As shown, the dominant therapeutic areas are represented by anti-leukemia, anti-breast cancer, and anti-lung cancer medications, where 18 related new drugs have been approved for the market. Anti-prostate cancer drugs received three approvals. Moreover, two anti-melanoma drugs, two anti-GIST drugs, three anti-lymphoma agents, two anti-myeloma drugs, and two anti-ovarian cancer drugs were also approved for the market. In addition, ten other anti-tumor agents were also released into the market for the targeted treatment of cancer. The information pertaining to the drugs approved between 2016 and 2020, including drug name, the research and development company, active ingredients, approval dates, and clinical applications, is summarized in Table 1.

Approved anti-cancer drugs (small-molecule) from 2016 to 2020.
Illustrative Compilation of Approved Anti-Cancer Drugs (Small-Molecule) From 2016 to 2020, Including Drug Name, Research and Development Company, Active Ingredients, Target, Approval Date, and Clinical Applications.

Abbreviations: ALK, anaplastic lymphoma kinase; AML, acute myeloid leukemia; GIST, gastrointestinal stromal tumor; NSCLC, non-small cell lung cancer; RRMM, relapsed or refractory multiple myeloma.
Furthermore, to provide insight into next-generation drug discovery, the medicinal chemistry synthetic routes and large-scale synthetic methods of 42 approved anti-cancer small-molecule drugs are discussed in this review. Chemical structure analysis and synthetic route discussion are beneficial for the contemporary medicinal chemistry strategies of next-generation drug discovery.
Gilteritinib (I)
Gilteritinib (I), a dual FMS-like tyrosine kinase 3 (FLT3)/AXL inhibitor, was co-developed by Astellas and Kotobuki Pharmaceuticals. It was approved in November 2018 to treat adult patients with acute myeloid leukemia (AML) displaying a mutation in the FLT3 gene, relapsed AML, or who have shown no improvement after previous treatment. 2
The synthesis of giltertinib (I), as described in a Chinese patent, is depicted in Scheme 1. The starting material, 3-bromo-5-chloro-6-ethylpyrazine-2-carbonitrile (

Synthesis of gilteritinib (I).
Owning to its critical role in various cancers, the Hedgehog signaling pathway has attracted significant attention as a potential avenue for developing new oncological therapies.4,5 Therefore, Pfizer has developed glasdegib (II), the first Hedgehog pathway inhibitor, to treat adult patients with newly-diagnosed AML, who are at least 75 years old and precluded from the use of standard chemotherapy. 6 It has been demonstrated that glasdegib (II) binds to the transmembrane protein, Smoothened (SMO), to inhibit the Hedgehog pathway. Recent evidence suggests that several other drugs targeting this pathway have been approved for advanced basal-cell carcinoma and are now being investigated for other indications. 7
The synthetic route to glasdegib (II) is depicted in Scheme 2.
8
The starting material, 4-methoxypyridine (
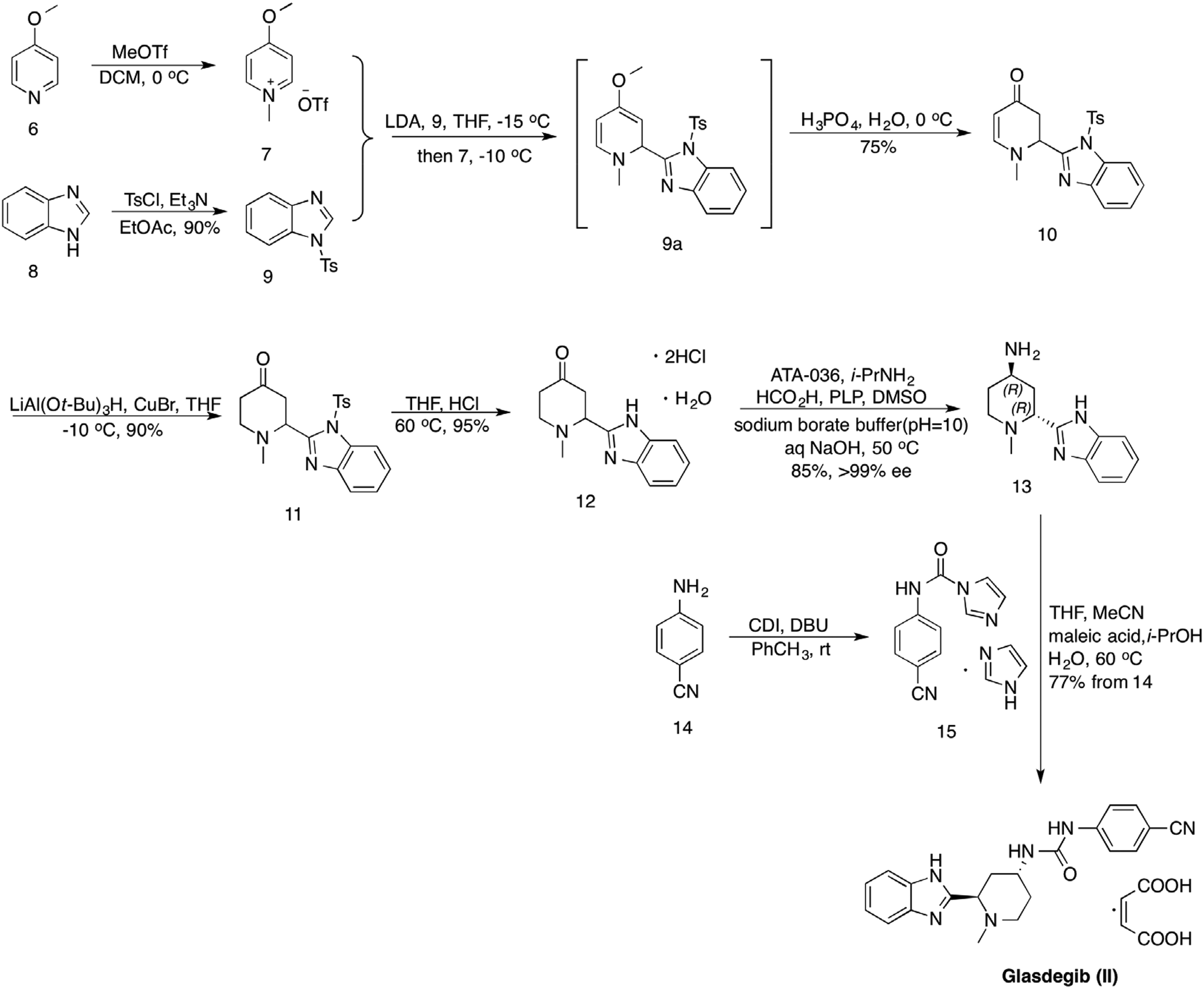
Synthesis of glasdegib (II).
Key intermediate
Due to its crucial role in the formation and progression of AML, gliomas, and other cancers, 2-hydroxyglutarate (2-HG), a metabolite produced from the mutated form of the IDH1 enzyme, has been identified as an important biomarker. Elevated 2-HG levels interfere with cellular metabolism and epigenetic regulation, contributing to oncogenesis. 9 Therefore, mIDH1 is considered a promising drug target for cancer treatment. Ivosidenib (III), a first-in-class orally available inhibitor developed by Agios Pharmaceuticals, was approved in July 2018 for treating adult patients with relapsed or refractory AML with a susceptible IDH1 mutation. 10 Ivosidenib (III) targets the IDH1 metabolic pathway to facilitate a profound oncometabolic 2-HG reduction in tumor models.7,11
The synthetic route to ivosidenib (III) is shown in Scheme 3.
12
First, a Schiff base

Synthesis of ivosidenib (III).
Similar to mIDH1, IDH2 is primarily located in mitochondria, mutating in many adult patients with AML. This increases 2-HG production to restrict cell differentiation, causing cells to lose their ability to advance from an immature to a fully differentiated state. However, IDH2 mutations are more common (8%-19% of patients) than mIDH1 (7%-14%) in adults with AML. Furthermore, the oncometabolite produced by mIDH2 is higher than mIDH1.13–15 Consequently, mIDH2 is considered a vital anti-cancer drug target. Enasidenib (IV) is a first-in-class, oral, selective mIDH2 inhibitor developed by Agios and later licensed to Celgene. It was approved in August 2017 for treating patients with relapsed or refractory AML with an IDH2 mutation.
The synthesis route of enasidenib (IV) was released by Agio in 2015
16
and involved seven steps described in Scheme 4. No yields were reported in the transformations. It began with the reaction between 2-(trifluoromethyl)pyridine (
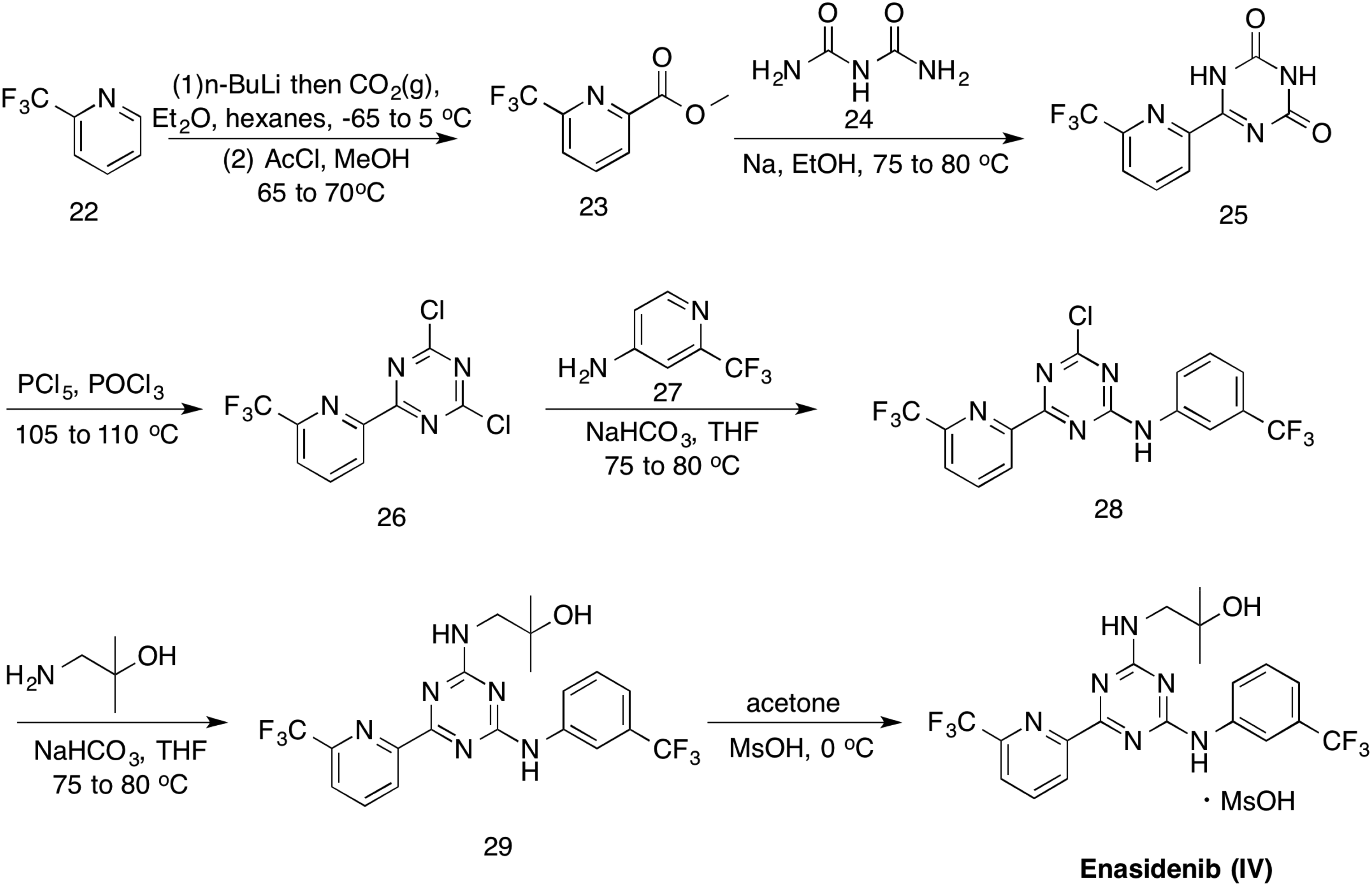
Synthesis of enasidenib (IV).
Midostaurin (V) is a natural multikinase inhibitor developed by Novartis Pharmaceuticals and was approved in April 2017 and combined with chemotherapy medications (daunorubicin and cytarabine) to treat adult patients with FLT3 mutation-positive AML, or aggressive systemic mastocytosis (SM) with associated blood cancer and mast-cell leukemia. Approximately 30% of adult patients suffering from AML with FLT3 mutations. Midostaurin (V) is the first multikinase inhibitor approved for the FLT3-mutant subtype.17–19
Midostaurin (V) was synthesized from the natural product staurosporine (

Synthesis of midostaurin (V).
B cell lymphoma subtype 2 (BCL-2) is often overexpressed in malignant cells, making this protein the rational target for anti-cancer therapy. 21 Venetoclax (VI) is a selective oral inhibitor of BCL-2 and was approved in April 2016 for treating chronic lymphocytic leukemia (CLL) in patients with 17p deletion who have received at least one prior therapy. The drug was co-developed by AbbVie and Genentech.
The synthetic route to venetoclax (VI) is depicted in Scheme 6.
22
The acquisition of the three key intermediates,

Synthesis of venetoclax (VI).
Moreover, intermediate
Tucatinib (VII)
Tucatinib (VII), also known as irbinitinib, ARRY-380, or ONT-380, is a selective, oral human epidermal growth factor receptor 2 (HER2) (also called ErbB-2) inhibitor developed by Array BioPharma and Seattle Genetics. 23 It was approved in April 2020 and combined with trastuzumab and capecitabine to treat adult patients with advanced, unresectable, or metastatic HER2-positive breast cancer and colorectal cancer. 24
Its synthetic route was developed by Array BioPharma Inc., as shown in Scheme 7.
25
Starting with picolinic acid (

Synthetic route 2 to tucatinib (VII).

Synthetic route 1 to tucatinib (VII).
Approximately 40% of patients with hormone receptor (HR)-positive, HER2-negative breast cancer reportedly have activated mutations in the PIK3CA gene, which encodes the catalytic (p110α) subunit of phosphatidylinositol 3-kinase (PI3 K). Therefore, targeting α-specific PI3 K provides a promising approach for anti-cancer therapy. Novartis Oncology has developed alpelisib as an orally bioavailable, α-specific PI3 K inhibitor, which was approved in May 2019 to treat advanced or metastatic breast cancer patients who are HR-positive and HER2-negative with PIK3CA mutations. Alpelisib (VIII) selectively inhibits p110α approximately 50 times more than other isoforms.26,27
The synthesis approach of alpelisib (VIII) is shown in Scheme 9.
28
Compound

Synthesis of alpelisib (VIII).
Polyadenosine 5′-diphosphoribose polymerase (PARP) plays a crucial role in repairing DNA single-strand breaks via the BER pathway. Therefore, as a promising drug target, inhibiting PARP results in an accumulation of DNA single-strand breaks, leading to cell death. 29 Talazoparib (IX) is an oral PARP inhibitor developed by Pfizer and approved in October 2018 to treat locally advanced or metastatic breast cancer patients with a germline BRCA mutation. 30
Scheme 10 shows the talazoparib (IX) synthetic route.
31
An aldol reaction between

Synthesis of talazoparib (IX).
Abemaciclib (X) is an oral inhibitor that targets cyclin-dependent kinases (CDK) 4 and 6. It was developed by Eli Lilly and approved in September 2017 for treating HR-positive and HER2-negative advanced breast cancer. 14 It is combined with fulvestrant when the breast cancer worsens after hormonal therapy or taken alone when the breast cancer worsens after hormonal therapy and chemotherapy.32,33
The synthesis of abemaciclib (X) is shown in Scheme 11.
34
Key intermediate

Synthesis of abemaciclib (X).
In July 2017, an oral, irreversible inhibitor of HER1 (EGFR), HER2, and HER4, known as neratinib (XI), was discovered by Wyeth and is currently being developed by Puma Biotechnology. 35 The drug was approved for treating early-stage HER2-positive breast cancer in women who have received previous trastuzumab and chemotherapy treatment as an adjuvant, as well as treating patients with HER2-positive early breast cancer at high risk of recurrence. 36
The synthesis of neratinib (XI) is described in Scheme 12.
37
4-Acetamido-3- ethoxyaniline (

Synthesis of neratinib (XI).
The overexpression of CDK4/6 is often evident in HR-positive breast cancer, while CDK4/6 inhibition can lead to cell cycle arrest. 38 Consequently, CDK4/6 represents promising drug targets for breast cancer treatment. Ribociclib (XII) is an oral CDK 4 and 6 inhibitor developed by Novartis and approved for treating advanced HR-positive and HER2-negative breast cancer in postmenopausal women.
Several synthetic routes to ribociclib (XII) have been described in various patents. One of the efficient approaches is shown in Scheme 13.
39
The reaction of 2,4-dichloropyrimidine (

Synthesis of ribociclib (XII).
Capmatinib (XIII)
Mesenchymal-epithelial transition (MET) is considered an attractive target for therapeutic blockade in lung cancer.40,41 Capmatinib (XIII), a highly selective, potent small-molecule MET inhibitor, was developed by Novartis and approved in May 2020 for treating metastatic non-small cell lung cancer (NSCLC) with MET exon 14 mutations.
The synthetic method for capmatinib (XIII) is shown in Scheme 14.
42
The chlorination of 4-bromo-3-fluorobenzoic acid (

Synthesis of capmatinib (XIII).
Lurbinectedin (XIV) is an oncogenic transcription inhibitor developed by Pharma Mar and approved in June 2020 for treating adult patients with metastatic small-cell lung cancer (SCLC) displaying disease progression on or after platinum-based chemotherapy. The drug covalently binds to DNA, causing DNA damage and apoptotic cell death.43,44
The total synthesis of lurbinectedin (XIV) is described in Scheme 15.
45
Protecting the amino group in the starting compound

Synthesis of lurbinectedin (XIV).
Pralsetinib (XV) is a selective rearranged during transfection (RET) inhibitor, developed by Blueprint Medicines Corporation and approved for treating NSCLC, and is a once-daily, oral, RET-targeted therapy. Approximately 1% to 2% of NSCLC patients display RET alterations. 46
The synthesis of pralsetinib (XV) is described in Scheme 16.
47
2,4-Dichloro-6-methylpyrimidine (

Synthesis of pralsetinib (XV).
Lorlatinib (XVI) is a potent, selective, brain-penetrating, third-generation anaplastic lymphoma kinase (ALK)/ROS1 tyrosine kinase inhibitor (TKI), developed by Pfizer and approved in November 2018 for treating ALK-positive NSCLC. 48
A convergent approach was designed to synthesize lorlatinib (XVI) as depicted in Scheme 17.
49
First, pyrazole

Synthesis of lorlatinib (XVI).
Dacomitinib (XVII) is an orally administered, small-molecule irreversible inhibitor of HER1 (EGFR), HER2, and HER4, developed by Pfizer and approved in September 2018 for treating patients with metastatic NSCLC with EGFR exon 19 deletion or exon 21 L858R substitution mutations. Unlike first-generation EGFR TKIs (gefitinib and erlotinib), dacomitinib, as a second-generation EGFR TKIs, was designed to overcome some of the issues related to the acquired resistance caused by first-generation agents.50,51
The dacomitinib (XVII) synthesis process was reported by Pfizer, as shown in Scheme 18.
52
The nitro group of

Synthesis of dacomitinib (XVII).
On April 28, 2017, brigatinib (XVIII), developed by Ariad Pharmaceuticals, was approved for treating patients with metastatic ALK-positive NSCLC who have progressed on or are crizotinib intolerant. It is a second-generation ALK inhibitor and is active in vitro against L1196 M, E1210 K, and G1202R ALK domain mutations, which may mediate acquired resistance to other ALK inhibitors.53,54
A synthetic route to brigatinib (XVIII) has been reported by Ariad Pharmaceuticals and is outlined in Scheme 19.
55
2-Iodoaniline (

Synthesis of brigatinib (XVIII).
Relugolix (XVX)
Relugolix (XVX) is an orally selective antagonist of the human gonadotropin-releasing hormone (GnRH) receptor, which was developed by Myovant Sciences, and approved in December 2020 to treat advanced prostate cancer. Relugolix (XVX) rapidly inhibits the pituitary release of the luteinizing hormone and FSH and has been shown to lower testosterone levels in multiple phase 1 and phase 2 studies. During the phase 3 trial, relugolix (XVX) achieved rapid and sustained suppression of testosterone levels, which was superior to the results obtained with leuprolide, with a 54% lower risk of major adverse cardiovascular events.56,57
One of the synthetic routes to relugolix (XVX) is described in Scheme 20.
58
Intermediate

Synthesis of relugolix (XVX).
Darolutamide (XX) is an androgen-receptor antagonist developed by Bayer and approved in July 2019 for treating adult patients with non-metastatic castration-resistant prostate cancer. Darolutamide (XX) inhibits AR variants, including W741L, T877A, H874Y, and F876L mutants. It competitively binds to lacking AR-ligand binding domains (LBD) with high binding affinity, significantly inhibiting the growth of enzalutamide-resistant prostate cancer cells in vivo. Compared with enzalutamide and apalutamide, darolutamide (XX) exhibited poor brain penetrance, decreasing seizure risk.59,60
An efficient large-scale approach has been reported by Orion Corporation, as shown in Scheme 21.
61
A coupling reaction between starting compound

Synthesis of darolutamide (XX).
Apalutamide (XXI) is a non-steroidal next-generation oral androgen receptor (AR) inhibitor developed by Janssen Biotech and was approved in 2018 for treating non-metastatic and castration-resistant prostate cancer. By binding to the LBD of the AR, apalutamide (XXI) blocks the effect of androgens, inhibiting nuclear translocation, DNA binding, and AR-mediated transcription.62,63
The synthetic route to apalutamide (XXI) was reported by Aragon.64,65 As shown in Scheme 22, the hydroxyl group of the starting material, 5-nitro-3-(trifluoromethyl)pyridin-2-ol (

Synthesis of apalutamide (XXI).
Encorafenib (XXII)
BRAFV600 mutations are observed in approximately 50% of melanomas. BRAFV600E mutation occurs in 35% to 50% of patients with melanoma. Combining a BRAF inhibitor with a MEK inhibitor simultaneously enhances efficacy and reduces toxicity.66,67 Encorafenib (XXII) is a BRAF inhibitor developed by Array BioPharma and approved in June 2018 in combination with binimetinib (XXIII) to treat unresectable or metastatic melanoma with a BRAFV600E or –V600K mutation.
The synthesis of encorafenib (XXII) is shown in Scheme 23.
68
Two intermediates (

Synthesis of encorafenib (XXII).
Binimetinib (XXIII), a MEK inhibitor, has been developed by Array BioPharma and was approved in June 2018 for treating unresectable or metastatic melanoma with a BRAFV600E or –V600K mutation. Binimetinib (XXIII) reversibly inhibited MEK1 and MEK2. Combining binimetinib (XXIII) with encorafenib (XXII) displayed more significant anti-proliferative activity toward BRAF-mutant cell lines than either drug alone.66,69
One of the synthetic routes to binimetinib (XXIII) is shown in Scheme 24.
70
23,4-Trifluoro-5-nitrobenzoic acid (

Synthesis of binimetinib (XXIII).
Avapritinib (XXIV)
Avapritinib (XXIV) is a potent and highly selective inhibitor of KIT and PDGFRα and was approved in 2020. Avapritinib (XXIV) was granted two breakthrough therapy designations, one for treating unresectable or metastatic GIST harboring the PDGFRα D842 V mutation and another for treating advanced SM, including the subtypes of aggressive SM, SM with an associated hematologic neoplasm, and mast-cell leukemia. 71
The synthesis of avapritinib (XXIV) was reported by Zhang in 2019 (Scheme 25).
72
The key intermediates,

Synthesis of avapritinib (XXIV).
Since mutations of the gene encoding receptor tyrosine kinases, KIT and platelet-derived growth factor receptor α (PDGFRA), have been found in more than 85% of GIST cases, KIT and PDGFRA have been considered promising targets for cancer treatment. Ripretinib (XXV) is a novel type II tyrosine switch control inhibitor developed by Deciphera Pharmaceuticals and was approved in May 2020 to treat adult patients with advanced GIST who have received prior treatment with ≥ 3 kinase inhibitors, including imatinib. It inhibits not only the wild-type KIT and PDGFRA kinase but also the secondary mutations. Furthermore, it inhibits other kinases, such as PDGFRB, TIE2, VEGFR2, and BRAF. 73
The preparation of ripretinib (XXV) and its fragments are described in Scheme 26.
74
Intermediate

Synthesis of eipretinib (XXV).
Then, a nitration reaction was conducted between
Zanubrutinib (XXVI)
Zanubrutinib (XXVI) is a potent and highly selective Bruton tyrosine kinase (BTK) inhibitor, developed by BeiGene, and was approved for treating adult patients with mantle-cell lymphoma (MCL) who have received at least one prior treatment for cancer.75,76
The synthetic route to zanubrutinib (XXVI) is described in Scheme 27.
77
First, acid
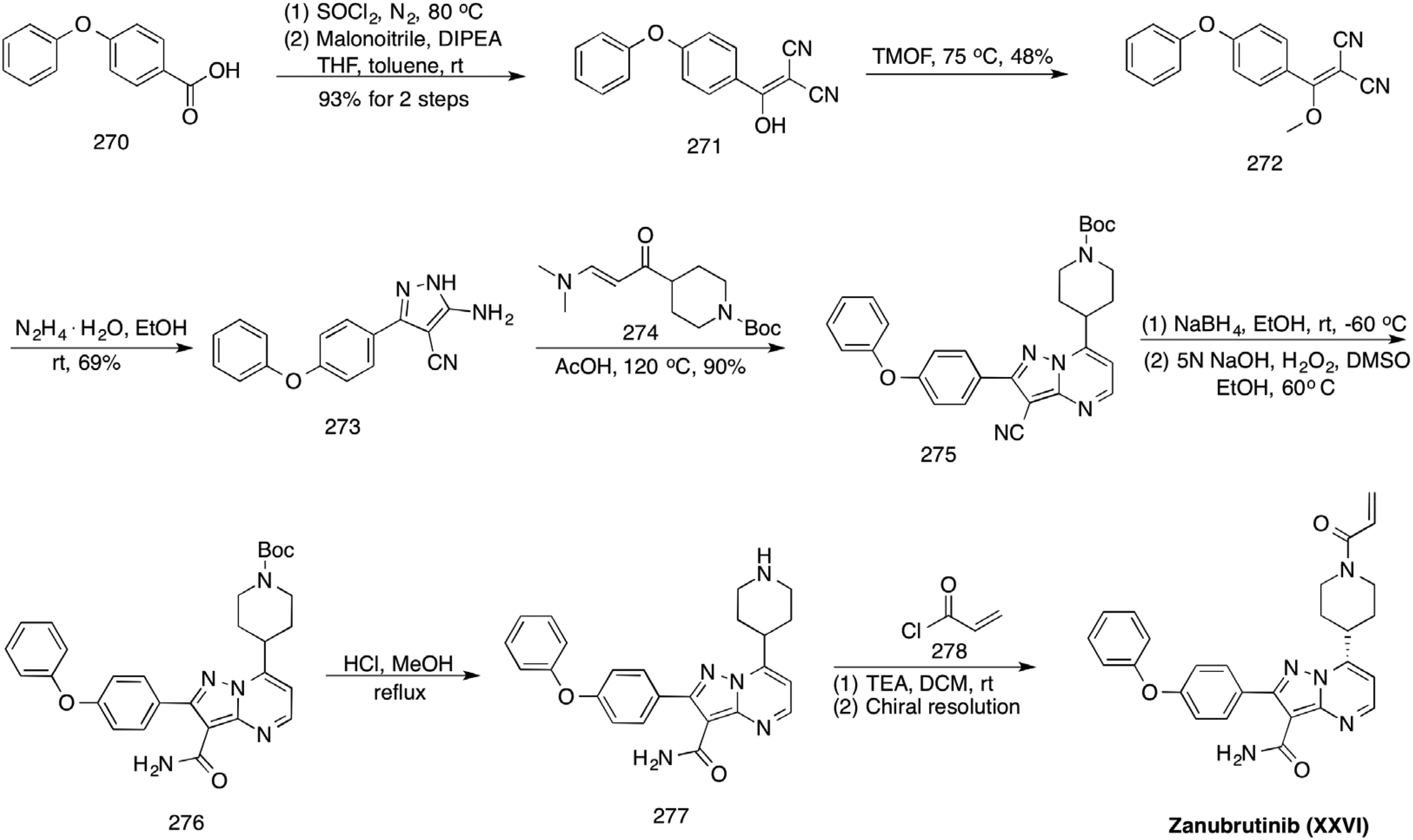
Synthesis of zanubrutinib (XXVI).
Developed by AstraZeneca Pharmaceuticals, acalabrutinib (XXVII) is a BTK inhibitor that was approved in October 2017 for treating adult patients with MCL who have received at least one prior cancer treatment. It is a second-generation BTK inhibitor that displays better selectivity and potency of ibrutinib, and it has received accelerated approval for MCL treatment.78,79
An efficient synthesis method for obtaining acalabrutinib (XXVII) is depicted in Scheme 28.80,81 First, POCl3 was used to prepare 4-bromobenzoyl chloride, which was then condensed with 2-aminopyridine to produce the precursor of

Synthesis of acalabrutinib (XXVII).
Follicular lymphoma is a slow-growing type of non-Hodgkin lymphoma. 82 Copanlisib (XXVIII), developed by Bayer, is a pan-class I PI3 K inhibitor. Copanlisib (XXVIII) exhibits excellent inhibitory activity against class I PI3 K α, β, γ, and δ isoforms with IC50 values of 0.5 nmol/L, 3.7 nmol/L, 6.4 nmol/L, and 0.7 nmol/L, respectively. It was approved to treat patients with relapsed follicular lymphoma who have received at least two prior systemic therapies.
The standard synthetic route to copanlisib (XXVIII) was released by Bayer.
83
As shown in Scheme 29, nitration of 4-formyl-2-methoxyphenyl acetate (

Synthesis of copanlisib (XXIII).
Fedratinib (XXIX)
Approximately 20% of patients with myelofibrosis have died from leukemic transformation, which is most commonly due to the V617F mutation in the JAK2 protein.84,85 Fedratinib (XXIX), developed by Celgene, is an oral JAK2-selective inhibitor that can inhibit STAT3/5 phosphorylation, reduce cell proliferation, and induce apoptosis in cells with JAK2 V6 17F or FLT3/ITD mutations. It was approved in August 2019 for treating intermediate-2 or high-risk primary or secondary myelofibrosis.
Scheme 30 shows the efficient synthetic route to fedratinib (XXIX).
86
First, a coupling reaction was carried out between compound 2-chloro-5-methylpyrimidin-4-amine and

Synthesis of fedratinib (XXIX).
As the major nuclear exporter of tumor suppressor proteins, the growth regulator and oncoprotein mRNAs, XPO1, are overexpressed in various cancer types and are identified as a promising drug target. Selinexor (XXX) is a first-in-class, oral, nuclear export protein exportin 1 (XPO1) inhibitor, which was developed by Karyopharm Therapeutics and was approved in July 2019 to treat adult patients with relapsed or refractory multiple myeloma (RRMM) in combination with dexamethasone. By forming a slowly reversible covalent bond with cysteine 528 in the binding pocket of XPO1, selinexor (XXX) selectively inhibits XPO1, leading to the accumulation of tumor suppressor proteins in the nucleus, decreasing oncoprotein levels, cell cycle arrest, and cancer cell apoptosis. 87
The synthesis of selinexor (XXX) was initiated with 3,5-bis(trifluoromethyl)benzonitrile (

Synthesis of selinexor (XXX).
Rucaparib (XXXI)
Rucaparib (XXXI), an oral, small-molecule poly (ADP-ribose) polymerase inhibitor, was developed by Clovis Oncology Inc. and approved for treating patients with deleterious BRCA mutations (germline and somatic) associated advanced ovarian cancer who have received two or more chemotherapy treatments. The drug inhibits PARP enzymes, including PARP-1, -2, and -3, which are integral in the DNA damage response system by activating the response pathways and facilitating repair.29,89
The most likely synthetic approach to rucaparib (XXXI) camsylate is described in Scheme 32.
90
Starting material

Synthesis of rucaparib (XXXI).
Niraparib (XXXII) is an oral, once-daily, highly-selective poly (ADP-ribose) polymerase (PARP)-1 and PARP-2 inhibitor, developed by Tesaro, approved for treating adult patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in complete or partial response to platinum-based chemotherapy. It displays a highly selective inhibition toward PARP-1 and PARP-2 (IC50 = 3.8 and 2.1 nmol/L, respectively), which represents a 100-fold higher selectivity than with other PARP-family members (PARP-3, v-PARP, and TANK-1). 91
The synthesis approach of niraparib (XXXII) is described in Scheme 33.
92
At first, Friedel-Crafts acylation was performed using succinic anhydride to prepare the acid intermediate, which was subsequently esterified with i-PrOH to obtain

Synthesis of niraparib (XXXII).
The key intermediate,
Pemigatinib (XXXIII)
Pemigatinib (XXXIII) is an oral fibroblast growth factor receptor (FGFR)1, FGFR2, and FGFR3 inhibitor, developed by Incyte Corporation and approved in April 2020 for treating adult patients with previously treated, unresectable, locally advanced, or metastatic cholangiocarcinoma, and an FGFR2 fusion or other rearrangements. 93 Approximately 10% to 16% FGFR2 alterations are present in patients with intrahepatic cholangiocarcinoma. Pemigatinib (XXXIII) is a novel, potent drug that selectively inhibits the functionality of altered FGFR2.94,95
Wu has reported the synthesis of pemigatinib (XXXIII),
96
as depicted in Scheme 34. After introducing the ethanamine side chain, the obtained
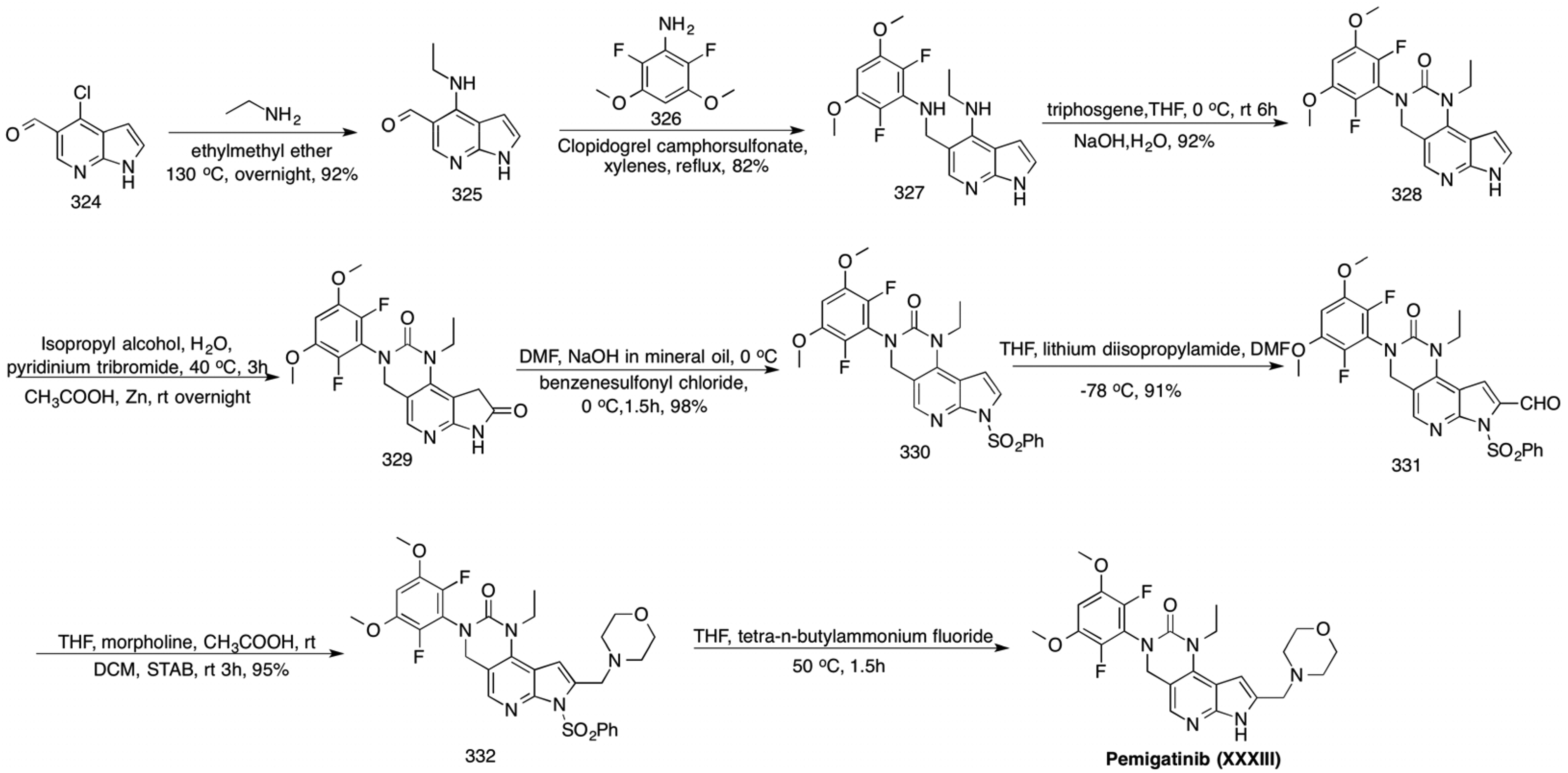
Synthesis of pemigatinib (XXXIII).
Tazemetostat (XXXIV), the first epigenetic drug for treating epithelioid sarcoma, was approved in 2020. It represents a new treatment strategy, especially for adults and pediatric patients aged 16 years and older with metastatic or locally advanced epithelioid sarcoma not eligible for complete resection. It was reported that, of the 62 participants with epithelioid sarcoma, about 10% experienced a reduction in their sarcoma after taking tazemetostat. Cancer proliferation ceased in more than 60% of the participants, even if their tumors did not shrink. What particularly stood out about the results was the duration of the benefit, lasting a year or more in many patients. The most common adverse reactions (incidence ≥20%) were pain, fatigue, nausea, decreased appetite, vomiting, and constipation. 97
The synthetic approach of tazemetostat (XXXIV) is shown in Scheme 35.
98
First, dibromatin was used to derive

Synthesis of tazemetosat (XXXIV).
Selumetinib (XXXV) is a potent, orally bioavailable, mitogen-activated protein kinase 1 and 2 (MEK1/2) inhibitor developed by AstraZeneca. It was approved in April 2020 for treating pediatric patients aged ≥ 2 years with neurofibromatosis type 1, who had symptomatic, inoperable plexiform neurofibromas. Selumetinib (XXXV) impedes RAS/RAF/MEK/ERK signaling, resulting in the antiproliferation and pro-apoptosis of tumor cells.99,100
According to the previously disclosed patent, the synthetic approach of selumetinib (XXXV) is shown in Scheme 36.
101
Nitration of 23,4-trifluorobenzoic (

Synthesis of selumetinib (XXXV).
Erdafitinib (XXXVI) is a pan-FGFR inhibitor developed by Janssen Pharmaceutical Companies. It was approved in April 2019 for treating locally advanced or metastatic, or unresectable urothelial carcinoma (mUC). By binding to FGFR, erdafitinib (XXXVI) inhibits FGFR phosphorylation and suppresses FGFR-related signal transduction pathways, resulting in tumor cell proliferation and death.102,103
The synthetic route to erdafitinib (XXXVI) was released by Astex Pharmaceuticals, as shown in Scheme 37.
104
The carbonyl group of starting material

Synthesis of erdafitinib (XXXVI).
Telotristat ethyl (XXXVII) is a first-in-class, small-molecule inhibitor of peripheral tryptophan hydroxylase (TPH) developed by Lexicon Pharmaceuticals and was approved in February 2017 for treating carcinoid syndrome diarrhea in combination with somatostatin analog (SSA) therapy in adults inadequately controlled by SSA therapy alone. Patients treated for carcinoid syndrome with telotristat extirpate (XXXVII) displayed reduced bowel movement (BM) frequency and decreased u5-HIAA without overt adverse CNS effects.105,106
Although several synthetic routes to telotristat ethyl (XXXVII) have been released, the most efficient has been reported by Lexicon, as shown in Scheme 38.
107
First, the key intermediate pinacol boronate

Synthesis of telotristat ethyl (XXXVII).
In the route to telotristat ethyl (XXXVII), the key intermediate
Pexidartinib (XXXVIII), developed by Daiichi Sankyo Inc., is an orally selective TKI against the colony-stimulating factor 1 (CSF1) receptor (IC50 = 0.02 μmol/L), KIT proto-oncogene receptor tyrosine kinase (KIT, IC50 = 0.01 μmol/L), and FLT3 harboring an internal tandem duplication mutation (FLT3-ITD, IC50 = 0.018 μmol/L). 108 The drug was approved in August 2019 for treating symptomatic tenosynovial giant-cell tumors (TGCT) associated with severe morbidity or functional limitations and not amenable to surgery. As a rare and locally aggressive neoplasm that overexpresses CSF1, TGCT is commonly found in the synovium of joints or tendon sheaths of young adults. 109
The synthesis approach of pexidartinib (XXXVIII) is described in Scheme 39.110,111 Treating

Synthesis of pexidartinib (XXXVIII).
Larotrectinib (XXXIX) is an oral, highly selective inhibitor of tropomyosin receptor kinase (TRK). It was developed by Bayer Health Care Pharmaceuticals Inc. and approved in November 2018 for treating adult and pediatric patients with solid tumors that display neurotrophic receptor tyrosine kinase (NTRK) gene fusions. Larotrectinib (XXXIX) is a first-generation TRK inhibitor that selectively targets pan-TRK (TRKA, TRKB, and TRKC) with half-maximal inhibitory concentration (IC50) values in the low nanomolar range, inhibiting the MAPK, PI3K–AKT, PKC, and STAT3 pathways.112,113
The synthesis of larotrectinib (XXXIX) is depicted in Scheme 40.
114
A Schiff base

Synthesis of larotrectinib (XXXIX).
Entrectinib (XXXX) is an orally selective ALK, ROS1, and TRKs inhibitor displaying nanomolar activity (ALK IC50 = 12 nM, ROS-1 IC50 = 7 nM, TRKA IC50 = 1 nM, TRKB IC50 = 3 nM, TRKC IC50 = 5 nM) on the corresponding target-driven cell lines. It was developed by Roche and was approved in June 2019 for treating adult and pediatric patients with NTRK fusion-positive, advanced or recurrent solid tumors.115,116
The synthetic route to entrectinib (XXXX) is described in Scheme 41.
117
At first,

Synthesis of entrectinib (XXXX).
Selpercatinib (XXXXI) is an ATP-competitive, highly selective receptor tyrosine kinase RET inhibitor. It was developed by Loxo Oncology and approved in May 2020 for treating adult patients with metastatic RET fusion-positive NSCLC, adult and pediatric patients ≥ 12 years of age with advanced or metastatic medullary thyroid cancer (MTC), and adult and pediatric patients ≥ 12 years of age with advanced or metastatic thyroid cancer who have received radioactive iodine that did not work or is no longer working.118,119
The synthetic route to selpercatinib (XXXXI) is depicted in Scheme 42.
120
At first, the hydroxylamine group was introduced into the 24,6-trimethylbenzenesulfonyl chloride (

Synthesis of selpercatinib (XXXXI).
Since the PI3 K signaling pathway plays an essential role in the activation, proliferation, and survival of B and T cells, PI3 K is considered a vital drug target. Duvelisib (XXXXII) is a first-in-class, small-molecule, selective dual inhibitor of PI3 K (PI3 K δ and PI3 K γ), which was initially created by Intellikine and further developed by Oncology (under the license of Infinity Pharmaceuticals). It was approved in September 2018 for treating adult patients with relapsed or refractory CLL/small lymphocytic lymphoma (SLL) after at least two prior therapies. Duvelisib (XXXXII) showed a 10-fold selectivity for PI3 K δ over PI3 K γ, while it was not active against other protein or lipid kinases.121,122
The synthesis of duvelisib (XXXXII) was released by Intellikine, as shown in Scheme 43.
123
Intermediate

Synthesis of duvelisib (XXXXII).
In the past five years, apart from these small molecules that have been approved for cancer treatment, several oligonucleotides, protein-based candidates, fusion protein, ADCs, and mAbs have also been marketed to fight for the tough battle. These non-small molecule drugs have disadvantages, such as the only medication route is intravenous medication, and these drugs are very expensive for the patients. However, small-molecule drugs have good oral bioavailability and are readily affordable.
The pharmaceutical community and academic researchers have made significant efforts to develop small-molecule drugs for fighting malignant tumors that severely threaten human health. Over the past five years, 42 small-molecule anti-cancer drugs have been approved for treating various types of cancer. Anti-lung cancer drugs, anti-breast cancer drugs, and anti-leukemia drugs account for almost half of these. These drugs act by targeting various proteins, enzymes, factors, and receptors, such as JAK3, EGFR, CDK-4 and 6, and PARP. The drugs summarized in this paper feature promising pharmacophores, which will benefit next-generation drug discovery.
In addition, the medicinal chemistry synthetic routes and large-scale synthetic methods of 42 approved anti-cancer small-molecule drugs are reviewed. In particular, metal-catalyzed coupling reactions (Suzuki coupling and the Buchwald-Hartwig reaction) are used to prepare drugs, such as abemaciclib (X), lorlatinib (XVI), and encorafenib (XXII). Cyclization and intramolecular or intermolecular cycloaddition represent the most important reactions employed to construct the pharmacophores, such as tucatinib (VII), neratinib (XI), ribociclib (XII), and lurbinectedin (XIV). To obtain the single enantiomer of the chiral drugs, asymmetric synthesis or chiral resolution are employed to synthesize several agents, such as ivosidenib (III), talazoparib (IX), darolutamide (XX), and avapritinib (XXIV). In addition, other reactions, such as Borch reduction and the Friedel-Crafts reaction, represent vital strategies for introducing side chains into the scaffold. Consequently, it is believed that the chemical structure analysis and synthetic route discussion are beneficial for contemporary medicinal chemistry strategies, including pharmacophore-based drug design, drug repositioning, lead diversification, multiparameter optimization, and drug discovery from natural resources.
Even having several advantages, small-molecule drugs for cancer treatment have several challenges, such as poor selectivity which leads to side effects. Also, tumor cells that long term exposed to a single small-molecule drug will develop drug resistance. In addition, some important antitumor targets, such as p53, KRAS, and STAT3, have been being called undruggable targets and have no small-molecule drugs approved till date. It was demonstrated that these proteins have no druggable pocket that small molecules can bind, which limited the development of small-molecule drugs. However, with the rapid development of chemical biology, new strategies such as PROTACs and molecular glues are the ideal solution to novel small-molecule antitumor drug discovery.
Footnotes
Acknowledgment
This project was supported by the Chun-Hui Project from the Ministry of Education of China (No. Z2017062), The Key Research and Development Program of Chengdu (Grant No. 2019-YF05-02095-SN), College Students' innovation and entrepreneurship training program (S202010650082).
Conflicts of Interest
These authors declare no conflict of interest.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the College Students' innovation and entrepreneurship training program (grant number S202010650082, No. Z2017062, No. 2019-YF05-02095-SN).
Ethical Approval
Not applicable, because this article does not contain any studies with human or animal subjects.
Informed Consent
Not applicable, because this article does not contain any studies with human or animal subjects.
Trial Registration
Not applicable, because this article does not contain any clinical trials.