
Abstract
Phenolic acid-polysaccharide conjugates, produced in plant food and medicine processing, are thought to account for the α-glucosidase inhibitory activity of the final products. However, this speculation lacks experimental support because of the complexity of the plant system and the polysaccharide structure. In this study, with dextran (average molecular weight, 1000) as the skeleton, a gallic acid-dextran conjugate was synthesized and confirmed by ultraviolet, infrared, and nuclear magnetic resonance spectroscopic analysis for the first time. Furthermore, this gallic acid-dextran conjugate showed inhibition of α-glucosidase due to galloyl groups in a mixed competitive and noncompetitive inhibition mode, whose performance was superior to that of acarbose.
Phenolic acids are widespread in plants. They have many health-promoting abilities, such as antioxidant, anticancer, anti-inflammatory, hypoglycemic, and antihypertensive activities. 1 -3 For a long time, reports about phenolic acids have focused on their extraction and bioactivity assessment. 4,5 Recent studies indicated that most phenolic acids in plants are attached to cell wall polysaccharides by covalent bonds. 6 -8 During plant food and medicine production, thermal, microwave, steam explosion, enzymatic hydrolysis, and other intense treatments can lead to the decomposition of phenolic acid-polysaccharide polymers in the raw materials to form water-soluble conjugates, which have a significant influence on the quality and function of the final products. 9,10 For example, the steam flash explosion improved the antioxidant and antiproliferative activities of wheat barn extracts. 11 Compared with native starch, starch ferulate possessed lower viscosity, higher water-holding capacity, and less retrogradation. 12 At the same time, the bioactivity and stability of polyphenols can also be enhanced through either conjugation or encapsulation with polymers. 13,14 It is thought that these conjugates may inhibit the digestive enzymes (such as α-glucosidase and α-amylase), which was the reason for the hypoglycemic performance of the final products. However, due to the complexity of the plant system and polysaccharide structure, it is difficult to confirm this speculation based on the traditional strategy of “separation-structure identification-bioactivity assessment.” This study is the first one in which a gallic acid-dextran conjugate was synthesized with dextran (average molecular weight 1000) as the skeleton, and its α-glucosidase inhibitory activity was measured.
Results and Discussion
Gallic acid is sensitive to temperature and pH. Its phenolic hydroxyl groups can be damaged during violent reactions, causing a loss in the bioactivity of its conjugates. Therefore, a protection step for the hydroxyl groups was first introduced in the synthetic route (Figure 1). In order to improve the reaction efficiency, triacetyl gallic acid was converted into triacetyl gallic acid chloride. Then, triacetyl gallic acid-dextran conjugate could be synthesized. After removing the acetyl groups under mild conditions, the gallic acid-dextran conjugate could be obtained.

Synthetic route of gallic acid-dextran conjugate.
The formation of a gallic acid-dextran conjugate was confirmed by ultraviolet (UV), infrared (IR), and nuclear magnetic resonance (NMR) spectroscopic measurements. Because of the absence of unsaturated bonds, dextran failed to have characteristic peaks in the range of 200-400 nm in its UV spectrum. However, the maximum absorbance peak for gallic acid was at 265 nm due to its conjugated double bonds. For the gallic acid-dextran conjugate, its UV spectrum was close to that of gallic acid, but its maximum absorption peak was 256 nm. This indicated that gallic acid had successfully combined with dextran through covalent bonds, resulting in the shift of its maximum absorption peak.
In its IR spectrum, dextran exhibited main absorption bands at 3412 cm−1 (for O-H stretching vibration), 2924 cm−1 (for C-H stretching vibration), 1653 (O-H bending vibration), and 1156 and 1016 cm−1 (for C-H and C-O stretching vibration). Gallic acid showed characteristic absorption bands for a hydroxyl group (3286 cm−1), C=O group (1705 cm−1), aromatic nucleus (1618, 1541, and 1469 cm−1), and C–O stretching vibration (1026 cm−1). For the gallic acid-dextran conjugate, its IR spectrum mainly demonstrated the characteristics of dextran, but a new characteristic band (1731 cm−1) could be observed, suggesting that dextran and gallic acid were covalently bound by an ester bond. In the 13C-NMR spectrum of the gallic acid-dextran conjugate, signals at 147.15, 141.11, and 127.39 ppm corresponded to the carbons of the galloyl group. Some new C signals (96.06, 92.17, 73.06 ppm) appeared around the original C signals of dextran (97.85, 73.35, 71.80, 71.37, 70.15, 65.51 ppm), which can be attributed to the substitution effect caused by the galloyl group. Based on UV, IR, and NMR results, it was confirmed that the gallic acid-dextran conjugate was successfully synthesized.
The α-glucosidase inhibitory activities of the gallic acid-dextran conjugate and dextran were compared. The measurement was carried out using p-nitrophenyl-α-d-glucopyranoside (pNP-G) as the substrate. α-Glucosidase can hydrolyze pNP-G to pNP, which has a maximum absorbance at 405 nm. 15 Therefore, the α-glucosidase inhibitory activity of the sample can be evaluated based on the production of pNP. As shown in Figure 2, dextran failed to exhibit α-glucosidase inhibitory activity in the concentration range of 20-200 μg/mL. However, the gallic acid-dextran conjugate showed strong α-glucosidase inhibitory capacity (half-maximal inhibitory concentration [IC50] 68.0 µg/mL) in a concentration-dependent manner, which was higher than that of acarbose (IC50 1.57 mg/mL). In order to clarify the underlying mechanism, the inhibitory mode of the gallic acid-dextran conjugate was studied based on the measurement of reaction rates (V) at different substrate concentrations (S). Results showed that there was a good linear relationship between 1/V and 1/S (Figure 3). With the increasing of S, the maximum reaction rate gradually declined, while the Michaelis constant increased, suggesting that the inhibitory mode of the gallic acid-dextran conjugate was a mixed competitive and non-competitive inhibition, which was consistent with the inhibitory mode of tannin. 16 However, low-molecular-weight polyphenols (such as flavonoids and phenolic acids) are usually competitive α-glucosidase inhibitors. 17,18

α-Glucosidase inhibitory activities of dextran (●), gallic acid-dextran conjugate (■), and acarbose (▲).
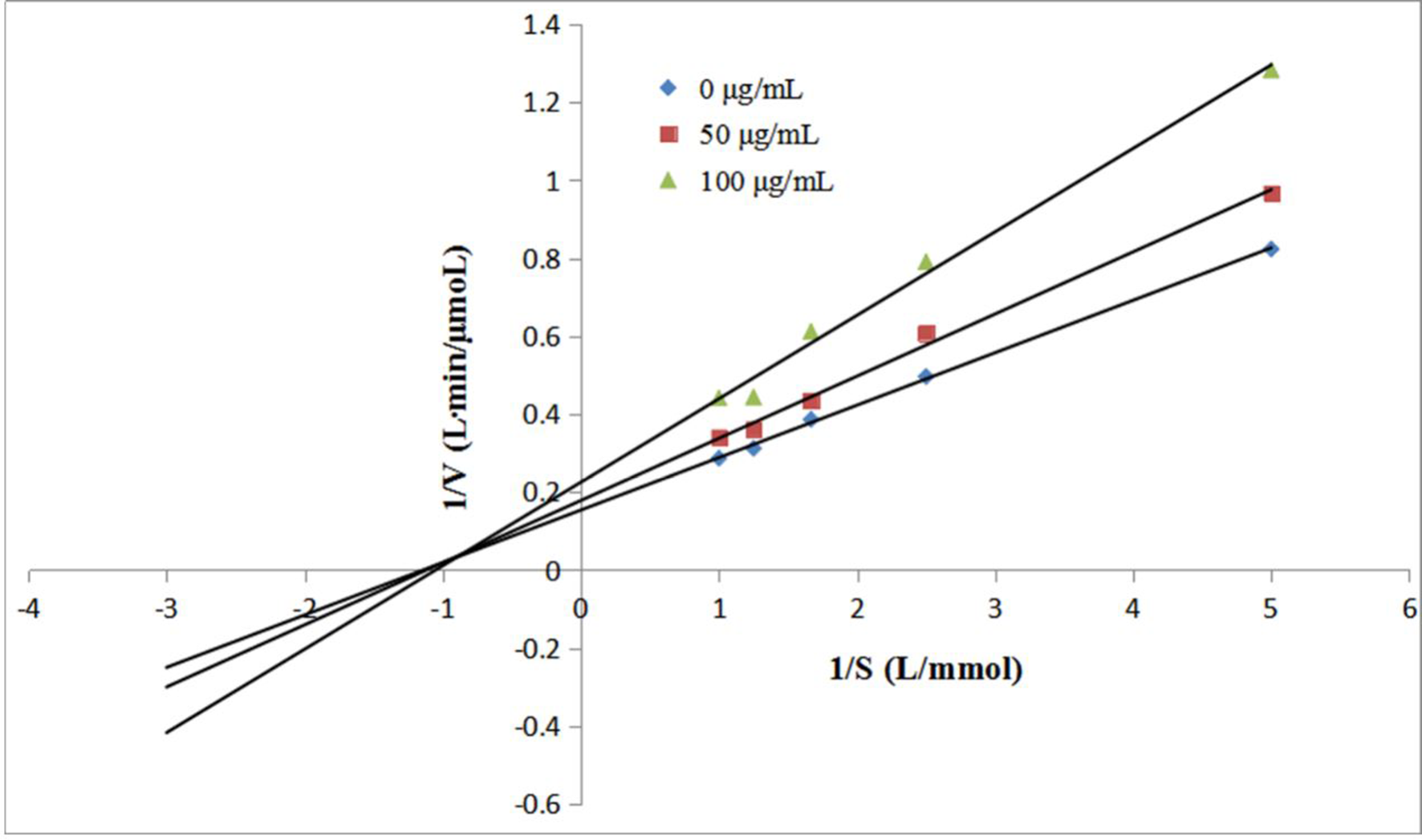
Lineweaver-Burk plot of gallic acid-dextran conjugate for determining the inhibitory mode.
The above results indicated the mechanism as follows: Dextran (average molecular weight <1000) has good water solubility. When it is connected with gallic acid by ester bonds, its hydrophobicity can be significantly improved. The obtained gallic acid-dextran conjugate is a tannin-like polyphenol, which can not only bind with the catalytic center of α-glucosidase to exert competitive inhibition but also combine with other hydrophobic sites to induce protein aggregation and accelerate enzyme loss.
Conclusions
In this study, a gallic acid-dextran conjugate was successfully synthesized for the first time. It exhibited strong α-glucosidase inhibitory activity in a concentration-dependent manner. Its inhibitory behavior was a mixed competitive and non-competitive inhibition. The obtained gallic acid-dextran conjugate can be used as a hypoglycemic nutraceutical in functional foods and medicines. Our results also confirmed that the phenolic acid-polysaccharide conjugates accounted for the α-glucosidase inhibitory activity of the final products, which was the reason why some processing treatments enhanced the bioactivities of plant foods and medicines.
Materials and Methods
Chemicals
Gallic acid, dextran (average molecular weight 1000), acetic anhydride, pyridine (anhydrous 99.8%), thionyl chloride, N,N-dimethylformamide, and carbon tetrachloride were purchased from Aladdin (Shanghai, China). α-Glucosidase from Saccharomyces cerevisiae and pNP-G were from Sigma-Aldrich (St. Louis, MO, USA). Ultrapure water was obtained using a Thermo GenPure UV/UF water system (Waltham, MA, USA). Other chemicals were of analytical grade.
Preparation of Gallic Acid-Dextran Conjugate
The gallic acid-dextran conjugate was synthesized according to the previous report, 12 which included the following steps.
Synthesis of triacetyl gallic acid
Gallic acid (15 g), acetic anhydride (35 mL), and anhydrous pyridine (30 mL) were mixed and stirred for 12 hours at room temperature. Afterward, 500 mL distilled water was added to the mixture, and an appropriate amount of hydrochloric acid was added to adjust the pH value to 1. The white precipitates were filtered, washed, dried under vacuum, and collected as triacetyl gallic acid (14.5 g).
Synthesis of triacetyl gallic acid chloride
Triacetyl gallic acid (10 g) and thionyl chloride (7.4 mL) were mixed and kept at 60 °C for 3 hours with N,N-dimethylformamide (0.02 mL) as the catalyst. The mixture was dried at 50 °C under vacuum. The obtained product was added to 75 mL carbon tetrachloride, incubated at 70 °C for 2 hours, and filtered. The supernatant was kept at 4 °C overnight. Then, the precipitated crystals were collected as triacetyl gallic acid chloride (6.52 g).
Synthesis of triacetyl gallic acid-dextran conjugate
Dextran (0.5 g; average molecular weight 1000) was dissolved in 10 mL of anhydrous pyridine. Then, 1 g of triacetyl gallic acid chloride was added. The mixture was heated at 120 °C for 4 hours. After cooling to room temperature, 200 mL of cold ethanol was added to the reaction system, the precipitate was filtered, dried, and collected as triacetyl gallic acid-dextran conjugate (0.30 g).
Synthesis of gallic acid-dextran conjugate
Triacetyl gallic acid-dextran conjugate (0.3 g) was mixed with 20 mL of saturated sodium bicarbonate ethanol solution, stirred for 2 hours at room temperature and filtered. The precipitate was vacuum-dried and collected as gallic acid-dextran conjugate (0.171 g) for the following experiments.
Structural Characterization
The UV spectra of the samples in the range of 200-400 nm were recorded on a Persee TU-1810 UV spectrophotometer (Beijing, China), the Fourier transformed-IR spectra on a Tensor 27 infrared spectrophotometer (Bruker, Germany) based on the potassium bromide method, and NMR measurements in deuterium oxide on an Avance 700 MHz NMR Spectrometer (Bruker, Germany).
α-Glucosidase Inhibitory Assay
The α-glucosidase inhibitory assay was carried out according to the previous report. 19 α-Glucosidase (0.1 U/mL), pNP-G (1 mM), dextran (20-200 μg/mL), and sample (20-200 μg/mL) solutions were prepared with 0.1 M of pH 6.8 phosphate buffer. α-Glucosidase solution (1 mL) and 0.5 mL of sample solution were mixed and incubated at 37 °C for 10 minutes. Then, the pNP-G solution (1 mL) was added. The above mixture was kept at 37 °C for 20 minutes. The reaction was terminated by adding 1 mL of ethanol. The absorbance of the mixture at 405 nm (A Sample) was measured. The absorbance of the control (A Control) containing phosphate buffer instead of the sample was also recorded. The α-glucosidase inhibitory activity could be determined based on the following equation:

Then, the reaction rates (V) at different substrate concentrations (S) were measured. The α-glucosidase inhibitory mode of the gallic acid-dextran conjugate was evaluated by drawing the plot of 1/V versus 1/S according to the Lineweaver-Burk method.
Footnotes
Acknowledgments
The authors are very thankful to all the authors whose work has been cited in this paper.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. 31771941), Key Scientific Research Project of Colleges and Universities in Henan Province of China (20zx016) and Open Project of National Engineering Laboratory for Wheat & Corn Further Processing of China (NL2018003).