
Abstract
As part of our ongoing project to search for natural product-based antiandrogens, nine derivatives of 2,3-dehydrosilybin have been synthesized for the evaluation of its antiproliferative activity in an androgen receptor-positive prostate cancer cell model. Specifically, 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin was synthesized through two approaches, and eight 23-O-substituted-3,5,7,20-O-tetramethyl-2,3-dehydrosilybins were achieved from 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin. The antiproliferative potency of 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin and its eight derivatives were assessed in an androgen receptor (AR)-positive LNCaP prostate cancer cell line, as well as in two AR-negative (PC-3 and DU145) prostate cancer cell models as a comparison. Our WST cell proliferation assay data indicate 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin and most of its 23-O-substituents can selectively inhibit AR-positive LNCaP prostate cancer cell proliferation. Our data suggest that 3,5,7,20-O-tetramethyl-2,3-dehydrosilibins could serve as a natural product-based scaffold for new antiandrogens for lethal castration-resistant prostate cancer.
Prostate cancer remains to be one of the big health concerns due to its high prevalence and mortality rate worldwide. Prostate cancer cases (191 930) that account for nearly 21% of all male cancer cases and 33 330 prostate cancer deaths are projected to occur in the United States in 2020. 1 Prostate cancer has been demonstrated to be driven by the androgen receptor (AR)-regulated gene expression that is initiated by the binding of androgens to the AR. 2 Androgen deprivation therapy (ADT) has thus been the therapeutic centerpiece for prostate cancer over 70 years. However, the median duration of ADT response is merely 18-24 months due to inevitably progression to castration-resistant prostate cancer (CRPC). 3 The major driving force for the continued progression of lethal CRPC is the reactivation of AR transcriptional activity. 4,5 The AR therefore continues to serve as the essential therapeutic target for CRPC. 2,3,6 The current Food and Drug Administration (FDA)-approved treatments for CRPC patients that target the AR-signaling axis include abiraterone acetate, 7 enzalutamide, 8 apalutamide, 9 and darolutamide. 10 Each of these treatments can only improve median overall survival by approximately 2-4 months. None is curative and treatment resistance is inevitable. 11,12 Novel drugs with a new structure scaffold targeting the AR-signaling axis are thus needed to treat deadly CRPC.
Silibinin (

Structures of silibinin and 2,3-dehydrosilybin.
Our previous studies on the optimization of silibinin as antiprostate cancer agents revealed: (1) that 2,3-dehydrosilybin (

Structure–activity relationships of 2,3-dehydrosilybins in prostate cancer cell models.

Antiproliferative potency of representative trimethyl-2,3-dehydrosilybins in prostate cancer cell models.
Results and Discussion
This study started with the preparation of 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin (

Biomimetic approach to the synthesis of tetramethyl-2,3-dehydrosilybin (7).
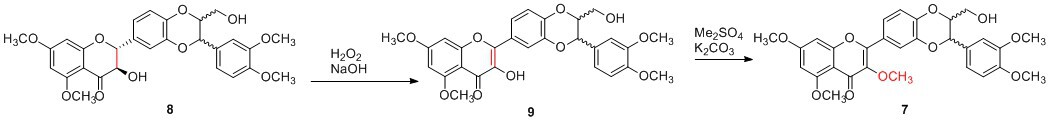
Semisynthesis of tetramethyl-2,3-dehydrosilybin (7).
With the critical intermediate 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin (

Synthesis of 23-O-substituted-2,3-dehydrosilybins (10-14).

Synthesis of 23-O-substituted-2,3-dehydrosilybins (15-17).
3,5,7,20-O-Tetramethyl-2,3-dehydrosilybin (
72 Hours of Antiproliferative Activity of 23-O-Substituted-2,3-Dehydrosilbinins.

aThe data were published in our previous paper. 25
Conclusion
In conclusion, this study demonstrated the effect of chemical modifications of the alcoholic hydroxyl group at C-23 of tetramethyl-2,3-dehydrosilybin on the antiproliferative potency and selectivity in AR-positive vs AR-negative prostate cancer cell models. 3,5,7,20-O-Tetramethyl-2,3-dehydrosilybin (
Experimental
Infrared (IR) spectra were recorded on a Nicolet Nexus 470 FTIR spectrophotometer. High-resolution mass spectra (HRMS)were obtained on a Q-Exactive Orbitrap mass spectrometer with electrospray ionization (ESI). Nuclear magnetic resonance (NMR) spectra were obtained on a Bruker Fourier 300 spectrometer in deuterated chloroform (CDCl3). The chemical shifts are given in ppm referenced to the respective solvent peak, and coupling constants are reported in hertz. Anhydrous tetrahydrofuran (THF) and dichloromethane (DCM) were purified by PureSolv MD7 Solvent Purification System from Innovative Technologies (MB-SPS-800). All other reagents and solvents were purchased from commercial sources and were used as is. Silica gel column chromatography was performed using silica gel (32-63 µM). Preparative thin-layer chromatography (PTLC) separations were carried out on TLC plates loaded with silica gel 60 GF254 (EMD Millipore Corporation). The diastereomeric silibinin (>98%) was purchased from Fisher Scientific (TCI America, CAS: 36804-17-8). Coniferyl alcohol was synthesized using the reported procedures.
35
The purity of compounds
Synthesis of 2,3-Dehydrosilbinin (2)
Quercetin dihydrate (1.67 g, 4.94 mmol, 1 eq) and coniferyl alcohol (1 g, 5.55 mmol, 1.1 eq) were dissolved in a mixture of toluene and acetone (300 mL in 2:1 ratio). The mixture was stirred for 10 minutes at 80°C prior to the addition of silver carbonate (1.53 g, 5.55 mmol, 1.1 eq). The reaction was allowed to proceed with stirring at 80°C overnight. The cooled reaction mixture was then filtered through a pad of silica gel; the filtrate was concentrated in vacuo. The crude product was sequentially purified through column chromatography (50% ethyl acetate in hexane) followed by PTLC (9% methanol [MeOH] in DCM, developed 3 times). The desired product (354 mg), obtained in 13% yield, still contains minor impurities but is good enough to be used for the next step reaction.
Rf: 0.33 (DCM–MeOH, 91:9, v/v).
1H NMR (300 MHz, CDCl3): 12.13 (s, 1 H, OH), 7.86 (s, 1 H, aromatic H), 7.85 (dd, J = 7.2, 2.1 Hz, 1 H, aromatic H), 7.17 (d, J = 1.8 Hz, 1 H, aromatic H), 7.08 (d, J = 9.3 Hz, 1 H, aromatic H), 7.01 (dd, J = 8.1, 1.8 Hz, 1 H, aromatic H), 6.95 (s, 1 H, aromatic H), 6.90 (d, J = 8.1 Hz, 1 H, OH), 6.58 (d, J = 2.1, 1 H, aromatic H), 6.27 (d, J = 2.1, 1 H, aromatic H), 5.05 (d, J = 8.1, 1 H, H-11), 4.25 (ddd, J = 7.8, 3.9, 2.4, 1 H, H-10), 3.89 (s, 3 H, OCH3), 3.79 (br.d, J = 12.6, 1 H, H-23), 3.55 (br.d, J = 13.5 Hz, 1 H, H-23).
The 1H NMR data of the obtained 2,3-dehydrosiliybinin are consistent with those reported in the literature. 32
Synthesis of 3,5,7,20-O-Tetramethyl-2,3-Dehydrosilybin (7)
Method 1
The suspension of 2,3-dehydrosilybin (
Method 2
Pure 5,7,20-O-trimethylsilybin (
MP: 193-195°C.
Rf: 0.62 (DCM–MeOH, 91:9).
IR (potassium bromide [KBr]): 3418, 2932. 2837, 1625, 1607, 1505 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.77-7.73 (m, 2 H, aromatic H), 7.08-7.05 (m, 2 H, aromatic H), 6.98 (d, J = 1.8 Hz, 1 H, aromatic H), 6.93 (d, J = 8.1 Hz, 1 H, aromatic H), 6.48 (d, J = 2.4 Hz, 1 H, aromatic H), 6.33 (d, J = 2.4 Hz, 1 H, aromatic H), 5.03 (d, J = 8.4, 1 H, H-11), 4.14 (dt, J = 8.1, 3.3, 1 H, H-10), 3.95 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3), 3.92 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 3.90-3.83 (overlapped, 1 H, H-23), 3.58 (d, J = 12.6 Hz, 1 H, H-23).
13C NMR (75 MHz, CDCl3): δ 174.3 (C-4), 164.1 (C-7), 161.1 (C-5), 158.9 (C-8a), 152.1 (C-16a), 150.0 (C-19), 149.6 (C-20), 145.3 (C-2), 143.8 (C-12a), 141.6 (C-3), 128.4 (C-17), 124.5 (C-14), 122.4 (C-15), 120.4 (C-22), 117.3 (C-13), 117.2 (C-16), 111.5 (C-21), 110.2 (C-18), 109.6 (C-4a), 95.9 (C-6), 92.4 (C-8), 78.8 (C-10), 76.5 (C-11), 61.9 (C-23), 60.1 (OCH3), 56.6 (OCH3), 56.2 (OCH3), 56.1 (OCH3), 56.0 (OCH3).
HRMS-ESI: m/z [M + H+] calcd for C29H29O10: 537.1761; found: 537.1761.
Synthesis of 3,5,7,20,23-O-Pentamethyl-2,3-Dehydrosilybin (10)
To a solution of 5,7,20-O-trimethyl-dehydrosilybin (
MP: 133-134°C.
Rf: 0.38 (DCM–MeOH, 95:5).
IR (KBr): 3490, 2930, 2838, 1621, 1604, 1579, 1506 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.76 (d, J = 2.1 Hz, 1 H, aromatic H), 7.73 (dd, J = 8.7, 2.1 Hz, 1 H, aromatic H), 7.10 (d, J = 8.4 Hz, 1 H, aromatic H), 7.02 (dd, J = 8.1, 1.8 Hz, 1 H, aromatic H), 6.96 (d, J = 1.8 Hz, 1 H, aromatic H), 6.93 (d, J = 8.1 Hz, 1 H, aromatic H), 6.47 (d, J = 2.4 Hz, 1 H, aromatic H), 6.32 (d, J = 2.1 Hz, 1 H, aromatic H), 5.05 (d, J = 8.1, 1 H, H-11), 4.18 (ddd, J = 8.1, 2.1, 1.8, Hz, 1 H, H-10), 3.94 (s, 3 H, OCH3), 3.92 (s, 6 H, 2 × OCH3), 3.90 (s, 3 H, OCH3), 3.86 (s, 3 H, OCH3), 3.62 (dd, J = 11.1, 2.1 Hz, 1 H, H-23). 3.36 (s, 3 H, OCH3), 2.23 (dd, J = 11.1, 3.9 Hz, 1 H, H-23).
13C NMR (75 MHz, CDCl3): 174.3 (C-4), 164.0 (C-7), 161.1 (C-5), 158.9 (C-8a), 152.2 (C-16a), 149.8 (C-19), 149.5 (C-20), 145.5 (C-2), 143.8 (C-12a), 141.5 (C-3), 128.8 (C-17), 124.3 (C-14), 122.3 (C-15), 120.3 (C-22), 117.5 (C-13), 117.2 (C-16), 111.4 (C-21), 110.3 (C-18), 109.6 (C-4a), 95.9 (C-6), 92.4 (C-8), 78.1 (C-10), 76.4 (C-11), 71.3 (C-23), 60.1 (OCH3), 59.7 (OCH3), 56.6 (OCH3), 56.2 (OCH3), 56.0 (OCH3), 55.9 (OCH3).
HRMS-ESI: m/z [M + H+] calcd for C30H31O10: 551.1917; found: 551.1916.
Syntheses of 3,5,7,20-O-Tetramethyl-23-Dimethylcarbamoyl-2,3-Dehydrosilybin (11), 3,5,7,20-O-Tetramethyl-23-Dimethylythiocarbamoyl-2,3-Dehydrosilybin (12), and 23-Diethylcarbamoyl-3,5,7,20-O-Tetramethyl-2,3-Dehydrosilybin (13)
To a solution of 3,5,7,20-O-tetramethyl-2,3-dehydrosilybin (
Derivative
Yield: 43%.
MP: 103-105°C.
Rf: 0.43 (DCM–MeOH, 95:5).
IR (KBr): 2931, 1702, 1625, 1605, 1505 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.77 (s, 1 H, aromatic H), 7.76 (dd, J = 10.5, 2.1 Hz, 1 H, aromatic H), 7.09 (d, J = 8.4 Hz, 1 H, aromatic H), 6.99 (dd, J = 8.1, 1.8 Hz, 1 H, aromatic H), 6.93 (d, J = 1.5 Hz, 1 H, aromatic H), 6.91 (d, J = 5.1 Hz, 1 H, aromatic H), 6.48 (d, J = 2.4 Hz, 1 H, aromatic H), 6.33 (d, J = 2.1 Hz, 1 H, aromatic H), 4.95 (d, J = 8.1 Hz, 1 H, H-11), 4.41-4.33 (m, 2 H, H-10, H-23), 4.03-4.00 (m, 1 H, H-23), 3.96 (s, 3 H, OCH3), 3.91 (s, 6 H, 2 × OCH3), 3.90 (s, 3 H, OCH3), 3.88 (s, 3 H, OCH3), 2.90 (s, 6 H, 2 × NCH3).
13C NMR (75 MHz, CDCl3): δ 174.0 (C-4), 164.0 (C-7), 161.2 (C-5), 158.9 (C-8a), 155.9 (carbamoyl carbon), 152.1 (C-16a), 150.1 (C-19), 149.7 (C-20), 145.2 (C-2), 143.8 (C-12a), 143.7 (C-3), 128.1 (C-17), 124.4 (C-14), 122.5 (C-15), 120.4 (C-22), 117.5 (C-13), 117.4 (C-16), 111.5 (C-21), 110.2 (C-18), 110.1 (C-4a), 96.0 (C-6), 92.5 (C-8), 77.0 (C-10), 76.5 (C-11), 63.9 (C-23), 60.2 (OCH3), 56.7 (OCH3), 56.6 (OCH3), 56.14 (OCH3), 56.10 (OCH3), 36.7 (NCH3), 36.1 (NCH3).
HRMS-ESI: m/z [M + H+] calcd for C32H34NO11: 608.2132; found: 608.2131.
Derivative
Yield: 44%.
MP: 111-113°C.
Rf: 0.50 (DCM–MeOH, 95:5).
IR (KBr): 2935, 2838, 1627, 1606, 1506 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.77 (s, 1 H, aromatic H), 7.76 (dd, J = 10.2, 2.4 Hz, 1 H, aromatic H), 7.08 (d, J = 9.0 Hz, 1 H, aromatic H), 7.00 (dd, J = 8.4, 1.8 Hz, 1 H, aromatic H), 6.93 (s, 1 H, aromatic H), 6.91 (d, J = 6.0 Hz, 1 H, aromatic H), 6.48 (d, J = 2.1 Hz, 1 H, aromatic H), 6.33 (d, J = 2.4 Hz, 1 H, aromatic H), 4.95 (d, J = 8.1 Hz, 1 H, H-11), 4.71 (dd, J = 12.0, 3.0 Hz, 1 H, H-23), 4.48 (ddd, J = 7.8, 4.5, 3.3 Hz, 1 H, H-10), 4.38 (dd, J = 11.7, 4.5 Hz, 1 H, H-23), 3.96 (s, 3 H, OCH3), 3.91 (s, 3 H, OCH3), 3.91 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 3.88 (s, 3 H, OCH3), 3.35 (s, 3 H, NCH3), 3.09 (s, 3 H, NCH3).
13C NMR (75 MHz, CDCl3): δ 187.4 (thiocarbamoyl carbon), 174.1 (C-4), 163.9 (C-7), 161.0 (C-5), 158.8 (C-8a), 151.9 (C-16a), 150.0 (C-19), 149.6 (C-20), 144.9 (C-2), 143.5 (C-12a), 141.4 (C-3), 127.8 (C-17), 124.4 (C-14), 122.4 (C-15), 120.2 (C-22), 120.0 (C-13), 117.2 (C-16), 117.1 (C-21), 111.4 (C-18), 109.9 (C-4a), 95.8 (C-6), 92.3 (C-8), 76.9 (C-10), 76.1 (C-11), 69.4 (C-23), 60.0 (OCH3), 56.4 (OCH3), 56.0 (2 × OCH3), 55.8 (OCH3), 42.9 (NCH3), 37.9 (NCH3).
HRMS-ESI: m/z [M + H+] calcd for C32H34NO10S: 624.1904; found: 624.1900.
Derivative
Yield: 40%.
MP: 95-96°C.
Rf: 0.37 (DCM–MeOH, 95:5).
IR (KBr): 2970, 2935, 2838, 2359, 2239, 1698, 1622, 1607, 1506 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.77 (s, 1 H, aromatic H), 7.75 (dd, J = 10.5, 2.1 Hz, 1 H, aromatic H), 7.08 (dd, J = 8.1, 0.9 Hz, 1 H, aromatic H), 6.98 (dd, J = 8.4, 1.8 Hz, 1 H, aromatic H), 6.93 (s, 1 H, aromatic H), 6.91 (d, J = 6.3 Hz, 1 H, aromatic H), 6.47 (d, J = 2.1 Hz, 1 H, aromatic H), 6.33 (d, J = 2.1 Hz, 1 H, aromatic H), 4.94 (d, J = 7.8 Hz, 1 H, H-11), 4.39–4.33 (overlapped, 2 H, H-10, H-23), 4.05-4.00 (m, 1 H, H-23), 3.95 (s, 3 H, OCH3), 3.91 (s, 6 H, 2 × OCH3), 3.90 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 3.35-3.18 (m, 4 H, 2 x NCH2 CH3), 1.14 (t, J = 7.2, 6 H, 2 x NCH2 CH3 ).
13C NMR (75 MHz, CDCl3): δ 174.2 (C-4), 164.0 (C-7), 161.2 (C-5), 158.9 (C-8a), 155.4 (carbamoyl carbon), 152.2 (C-16a), 150.1 (C-19), 149.7 (C-20), 145.2 (C-2), 143.7 (C-12a), 141.6 (C-3), 128.2 (C-17), 124.4 (C-14), 122.5 (C-15), 120.3 (C-22), 117.4 (C-13), 117.3 (C-16), 111.5 (C-21), 110.2 (C-18), 109.6 (C-4a), 96.0 (C-6), 92.5 (C-8), 77.0 (C-10), 76.6 (C-11), 63.6 (C-23), 60.1 (OCH3), 56.6 (OCH3), 56.2 (OCH3), 56.1 (OCH3), 55.9 (OCH3), 42.0 (NCH2 CH3 ), 41.5 (NCH2 CH3), 14.3 (NCH2 CH3 ), 13.6 (NCH2 CH3 ).
HRMS-ESI: m/z [M + H+] calcd for C34H38NO11: 636.2445; found: 636.2441.
Syntheses of 3,5,7,20-O-Tetramethyl-23-O-Methanesulfonyl-2,3-Dehydrosilybin (14) and 3,5,7,20-O-Tetramethyl-23-O-Toluenesulfonyl-2,3-Dehydrosilybin (15)
To a solution of
Compound
MP: 173-175°C.
Rf: 0.45 (DCM–MeOH, 95:5).
IR (KBr): 2936, 2839, 1625, 1606, 1518, 1506 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.78 (s, 1 H, aromatic H), 7.77 (dd, J = 10.2, 2.1 Hz, 1 H, aromatic H), 7.07 (d, J = 8.7 Hz, 1 H, aromatic H), 7.03 (dd, J = 8.4, 2.1 Hz, 1 H, aromatic H), 6.96 (s, 1 H, aromatic H), 6.95 (d, J = 10.5 Hz, 1 H, aromatic H), 6.48 (d, J = 2.1 Hz, 1 H, aromatic H), 6.34 (d, J = 2.4 Hz, 1 H, aromatic H), 4.99 (d, J = 8.4 Hz, 1 H, H-11), 4.46 (dd, J = 11.7, 2.7 Hz, 1 H, H-23), 4.35 (dt, J = 8.1, 2.7 Hz, 1 H, H-10), 4.14 (dd, J = 11.7, 3.9 Hz, 1 H, H-23), 3.96 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3), 3.92 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 3.88 (s, 3 H, OCH3), 3.10 (s, 3 H, SO2CH3).
13C NMR (75 MHz dimethylsulfoxide [DMSO]-d6 ): 174.2 (C-4), 164.1 (C-7), 161.2 (C-5), 158.9 (C-8a), 151.9 (C-16a), 150.3 (C-19), 149.8 (C-20), 144.6 (C-2), 143.6 (C-12a), 141.7 (C-3), 127.3 (C-17), 125.0 (C-14), 122.7 (C-15), 120.3 (C-22), 117.5 (C-13), 117.2 (C-16), 111.6 (C-21), 110.2 (C-18 & C-4a), 96.0 (C-6), 92.5 (C-8), 76.2 (C-10), 75.9 (C-11), 68.1 (C-23), 60.1 (OCH3), 56.6 (OCH3), 56.24 (OCH3), 56.20 (OCH3), 55.9 (OCH3), 37.9 (SO2CH3).
HRMS-ESI: m/z [M + H+] calcd for C30H31O12S: 615.1537; found: 615.1532.
Compound
Yield: 65%.
MP: 165-166 °C.
Rf: 0.57 (DCM–MeOH, 95:5).
IR (KBr): 3004, 2937, 2839, 2239, 1625, 1605, 1505 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.76 (d, J = 8.4 Hz, 2 H, aromatic H), 7.73-7.69 (overlapped, 2 H, aromatic H), 7.34 (d, J = 7.8 Hz, 2 H, aromatic H), 6.96-6.86 (overlapped, 4 H, aromatic H), 6.46 (d, J = 2.4 Hz, 1 H, aromatic H), 6.32 (d, J = 2.1 Hz, 1 H, aromatic H), 4.94 (d, J = 8.1 Hz, 1 H, H-11), 4.26-4.19 (m, 2 H, H-10, H-23), 4.02-3.99 (m, 1 H, H-23), 3.94 (s, 3 H, OCH3), 3.91 (s, 6 H, 2 × OCH3), 3.88 (s, 3 H, OCH3), 3.86 (s, 3 H, OCH3), 2.46 (s, 3 H, Ar-CH3).
13C NMR (75 MHz, CDCl3): δ 174.2 (C-4), 164.1 (C-7), 161.1 (C-5), 158.9 (C-8a), 151.9 (C-16a), 150.1 (C-19), 149.7 (C-20), 145.3 (C-2), 144.7 (toluenesulfonyl), 143.4 (C-12a), 141.6 (C-3), 132.6 (toluenesulfonyl), 130.0 (toluenesulfonyl), 128.2 (C-17), 127.4 (toluenesulfonyl), 124.6 (C-14), 122.5 (C-15), 120.2 (C-22), 117.3 (C-13), 117.2 (C-16), 111.5 (C-21), 110.2 (C-18), 109.6 (C-4a), 95.9 (C-6), 92.4 (C-8), 76.0 (C-10), 75.7 (C-11), 68.2 (C-23), 60.1 (OCH3), 56.5 (OCH3), 56.1 (2 × OCH3), 55.9 (OCH3), 21.9 (Ar-CH3).
HRMS-ESI: m/z [M + H+] calcd for C36H35O12S: 691.1850; found: 691.1846.
Synthesis of 23-Azido-3,5,7,20-O-Tetramethyl-2,3-Dehydrosilybin (16)
To a solution of compound
MP: 92-93°C.
Rf: 0.50 (DCM–MeOH, 95:5).
IR (KBr): 3055, 2936, 2103, 1626, 1607, 1519, 1505 cm−1.
1H NMR (300 MHz, CDCl3): δ 7.78-7.74 (overlapped, 2 H, aromatic H), 7.11 (d, J = 8.7 Hz, 1 H, aromatic H), 7.01 (dd, J = 8.1, 1.8 Hz, 1 H, aromatic H), 6.94 (d, J = 2.1 Hz, 1 H, aromatic H), 6.93 (d, J = 8.4 Hz, 1 H, aromatic H), 6.48 (d, J = 2.1 Hz, 1 H, aromatic H), 6.33 (d, J = 2.1 Hz, 1 H, aromatic H), 4.98 (d, J = 8.1 Hz, 1 H, H-11), 4.26 (ddd, J = 8.1, 4.2, 2.7 Hz, 1 H, H-10), 3.95 (s, 3 H, OCH3), 3.93 (s, 3 H, OCH3), 3.92 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 3.88 (s, 3 H, OCH3), 3.60 (dd, J = 13.5, 2.7, 1 H, H-23), 3.18 (dd, J = 13.5, 4.2, 1 H, H-23).
13C NMR (75 MHz, CDCl3): δ 174.3 (C-4), 164.1 (C-7), 161.2 (C-5), 158.9 (C-8a), 152.0 (C-16a), 150.1 (C-19), 149.7 (C-20), 145.2 (C-2), 144.9 (C-12a), 143.7 (C-3), 127.9 (C-17), 124.7 (C-14), 122.5 (C-15), 120.3 (C-22), 117.4 (C-13), 117.3 (C-16), 111.5 (C-21), 110.1 (C-18), 109.6 (C-4a), 96.0 (C-6), 92.5 (C-8), 68.4 (C-10), 60.5 (C-11), 60.1 (OCH3), 56.6 (OCH3), 56.22 (OCH3), 56.19 (OCH3), 55.9 (OCH3), 51.2 (C-23).
HRMS-ESI: m/z [M + H+] calcd for C29H28N3O9: 562.1826; found: 562.1846.
Synthesis of 23-Amino-3,5,7,20-O-Tetramethyl-2,3-Dehydrosilybin (17)
To a solution of compound
MP: 112-115°C.
Rf: 0.53 (DCM–MeOH, 91:9).
IR (KBr): 3490, 2937, 1628, 1603, 1518, 1507 cm-=−1.
1H NMR (300 MHz, CD3COCD3): δ 7.72 (dd, J = 8.4, 2.1 Hz, 1 H, aromatic H), 7.69 (d, J = 1.5 Hz, 1 H, aromatic H), 7.15 (d, J = 2.1 Hz, 1 H, aromatic H), 7.08-7.00 (overlapped, 3 H, aromatic H), 6.78 (d, J = 2.4 Hz, 1 H, aromatic H), 6.44 (d, J = 2.1 Hz, 1 H, aromatic H), 5.18 (d, J = 7.8, 1 H, H-11), 4.48 (ddd, J = 7.8, 4.8, 3.3, 1 H, H-10), 3.94 (s, 3 H, OCH3), 3.90 (s, 3 H, OCH3), 3.86 (s, 3 H, OCH3), 3.85 (s, 3 H, OCH3), 3.85 (s, 3 H, OCH3), 3.47 (dd, J = 15.3, 3.0 Hz, 1 H, H-23), 3.27 (dd, J = 15.3, 4.8 Hz, 1 H, H-23), 2.88 (s, 2 H, NH2).
13C NMR (75 MHz, CD3COCD3): δ 173.3 (C-4), 165.1 (C-7), 162.0 (C-5), 159.7 (C-8a), 152.3 (C-16a), 150.9 (C-19), 150.5 (C-20), 146.8 (C-2), 145.0 (C-12a), 142.1 (C-3), 130.4 (C-17), 124.8 (C-14), 122.6 (C-15), 121.4 (C-22), 117.8 (C-13), 117.6 (C-16), 112.6 (C-21), 112.4 (C-18), 110.2 (C-4a), 96.6 (C-6), 93.7 (C-8), 79.3 (C-10), 78.2 (C-11), 59.9 (OCH3), 56.5 (OCH3), 56.4 (OCH3), 56.3 (OCH3), 56.2 (OCH3), 52.5 (C-23).
HRMS-ESI: m/z [M + H+] calcd for C29H30NO9: 536.1921; found: 536.1907.
Cell Culture
All cell lines were initially purchased from the American Type Culture Collection. The PC-3 and LNCaP prostate cancer cell lines were routinely cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Cultures were maintained in a high humidity environment supplemented with 5% carbon dioxide at a temperature of 37°C. The DU145 prostate cancer cells were routinely cultured in Eagle’s Minimum Essential Medium supplemented with 10% FBS and 1% penicillin/streptomycin.
WST-1 Cell Proliferation Assay
PC-3, DU145, or LNCaP cells were plated in 96-well plates at a density of 3200 cells each well in 200 µL of culture medium. The cells were then treated with synthesized derivatives or silybin (as a positive reference) separately at different doses (1 µM, 3.125 µM, 6.25 µM, 12.5 µM, and 25 µM for compounds
Statistical Analysis
All data are represented as the mean ± standard deviation for the number of experiments indicated. Other differences between treated and control groups were analyzed using the Student’s t-test. A P value <0.05 was considered statistically significant.
Supplemental Material
Supplementary material - Supplemental material for 23-O-Substituted-2,3-Dehydrosilybins Selectively Suppress Androgen Receptor-Positive LNCaP Prostate Cancer Cell Proliferation
Supplemental material, Supplementary material, for 23-O-Substituted-2,3-Dehydrosilybins Selectively Suppress Androgen Receptor-Positive LNCaP Prostate Cancer Cell Proliferation by Ziran Jiang, Arman Sekhon, Yogeshwari Oka, Guanglin Chen, Nagat Alrubati, Jasleen Kaur, Alexia Orozco, Qiang Zhang, Guangdi Wang and Qiao-Hong Chen in Natural Product Communications
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was financially supported by California State University (CSU)-Fresno, USA. The HRMS data were supported by the NIH RCMI program at Xavier University of Louisiana through Grant 2G12MD007595 (G. Wang). We are also grateful to the Undergraduate Research Grant program at CSU-Fresno and CSU-LSAMP program funded by NSF under grant # HRD-1302873, CSU Office of the Chancellor, and Fresno State for the funding to N. Alrubati.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.