
Abstract
A catalytic asymmetric intramolecular Darzens reaction of 2-halomalonate derivatives was developed for the enantioselective preparation of chiral building blocks for epoxide-containing natural products. Among the screened catalysts, some phase-transfer catalysts gave the desired epoxide in moderate enantioselectivity, albeit in low yield. The epoxide product would be useful as versatile chiral building blocks for natural product synthesis.
Keywords
Since its discovery over a century ago, 1 the Darzens reaction has been widely recognized as a reliable, robust method for constructing the epoxide functionality, which is ubiquitous in bioactive molecules 2 -4 and can be used as a highly reactive functionality in organic synthesis. 5 However, to our knowledge, the Darzens reaction is poorly studied compared with the corresponding aldol reaction 6 because the stereoselectivity in the Darzens reaction is complicated due to its reaction mechanism, which consists of aldol addition and subsequent epoxide formation. 7,8 Nevertheless, the Darzens reaction is very important for epoxide formation from carbonyl compounds, along with the Corey-Chaykovsky reaction. 9,10
Recently, our research group has been intrigued by the unique molecular structures of epoxide-containing bioactive natural products, such as epolactaene, 11 L-755,807, 12 fusarin C, 13 and euvesperin A 14 (Figure 1).
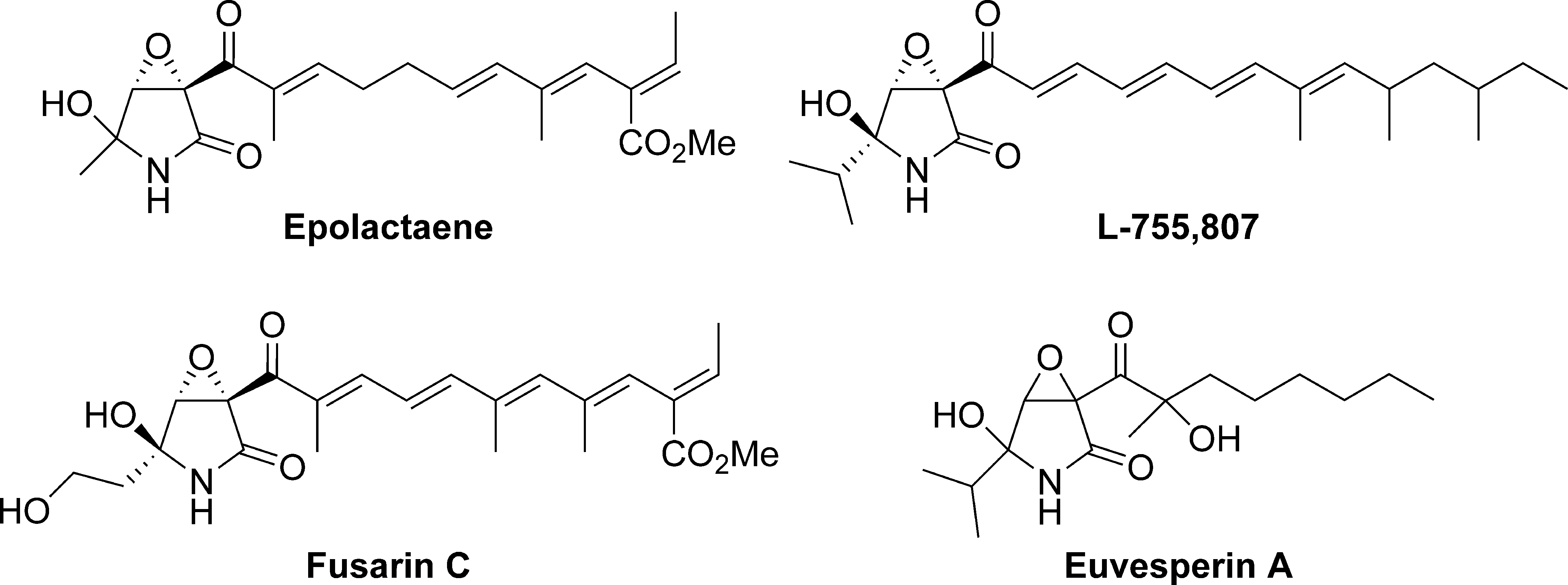
Structures of epolactaene, L-755,807, fusarin C, and euvesperin A.
During synthetic studies of these natural products, we developed a highly syn-selective Darzens reaction between optically active α-alkylsilyloxy aldehyde

syn-Selective Darzens reaction.
We envisioned that an enantioselective version of Darzens reaction would be more valuable in our system.
18
Thus, we investigated a catalytic asymmetric intramolecular Darzens reaction of 2-halomalonate derivative

Proposed asymmetric intramolecular Darzens reaction.
This paper describes the asymmetric intramolecular Darzens reaction of 2-halomalonate derivatives with chiral organocatalysts.
Initially, substrates
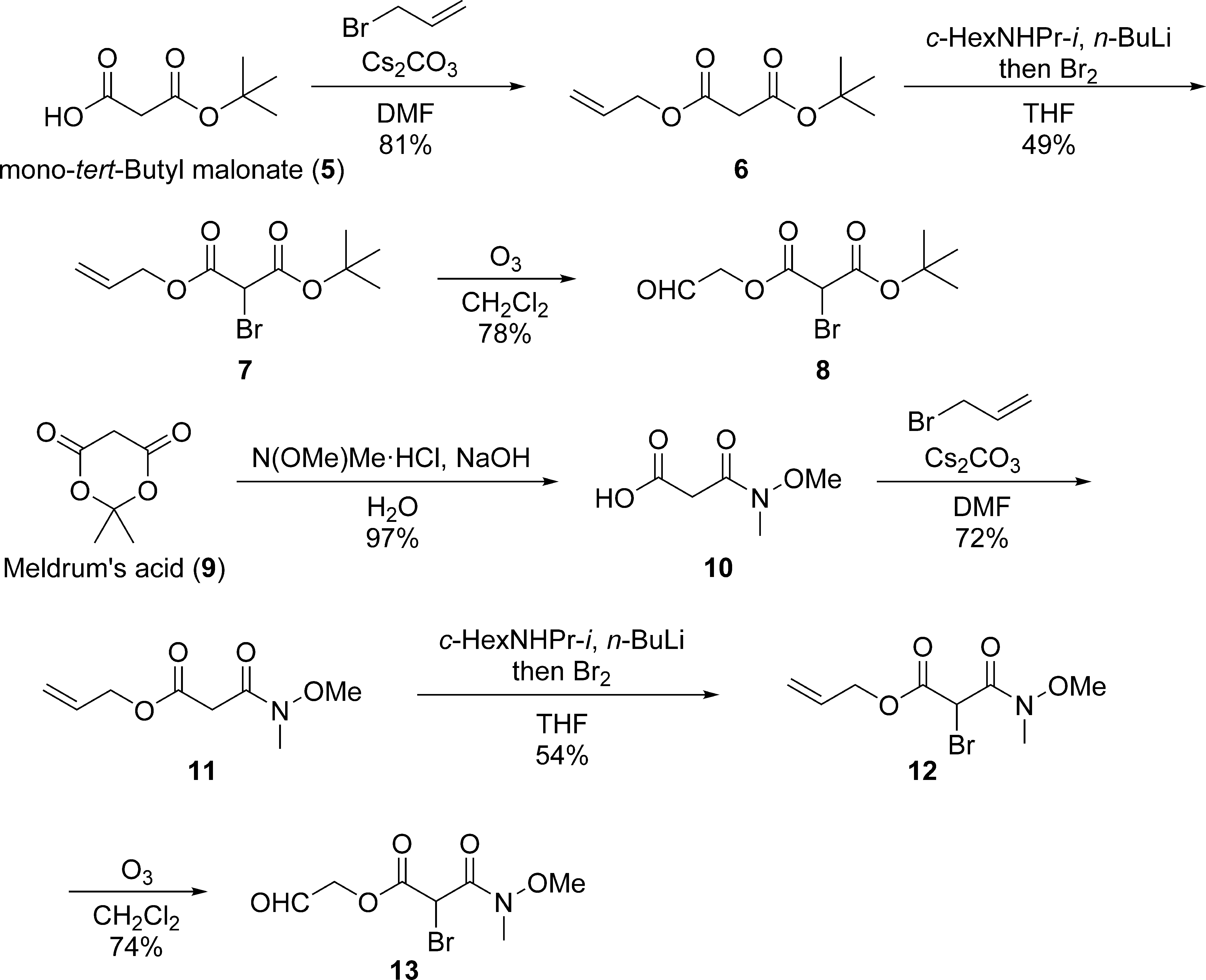
Synthesis of aldehydes 8 and 13 for intramolecular Darzens reaction.
Weinreb amide
With substrates
Intramolecular Darzens Reaction Using Organocatalysts.


aDetermined by chiral high-performance liquid chromatography.
bThe reaction was carried out at 0°C.
cThe reaction was carried out at –78°C.
With Weinreb amide
In conclusion, we demonstrated the intramolecular Darzens reaction of 2-halomalonate derivatives with chiral organocatalysts. The absolute configuration of the products and a plausible transition state for the present Darzens reaction will be reported in future work. The asymmetric induction was insufficient, although we believe this work is a promising starting point for development of a catalytic asymmetric intramolecular Darzens reaction. Structural optimization of the substrate to achieve high enantioselectivity is necessary, and this work is currently underway in our laboratory.
Experimental
1H NMR spectra were recorded on a JEOL JNM-AL300 (300 MHz) or JEOL JNM-ECS400 (400 MHz) spectrometer. The chemical shifts are expressed in ppm relative to tetramethylsilane (δ = 0) as an internal standard (CDCl3 solution). Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; and br, broad peak. 13C NMR spectra were recorded on a 100 MHz instrument (JNM-ECS400, JEOL). The chemical shifts are reported in ppm relative to the central line of the triplet at 77.0 ppm for CDCl3. Infrared (IR) spectra were measured on an IR spectrometer (VALOR-III, JASCO) and are reported in wavenumbers (cm−1). Both low-resolution mass spectra (MS) and high-resolution mass spectra (HRMS) were obtained using a double-focusing mass spectrometer (JMS 700, JEOL) in electron impact ionization (EI) or fast atom bombardment (FAB) mode with a direct inlet system. Column chromatography was performed on silica gel (40-100 mesh). Analytical thin-layer chromatography was conducted using 0.25 mm silica gel 60 F plates. Chiral high-performance liquid chromatography (HPLC) was performed on a Shimadzu HPLC with CHIRALPAK IC column (4.6 mmϕ × 250 mm, 5 µm particle size, 1.0 mL/min flow rate) equipped with a guard, employing a mixture of EtOH and n-hexane.
Allyl tert-Butyl Malonate (6)
Cesium carbonate (3.42 g, 10.5 mmol) and allyl bromide (0.90 mL, 11 mmol) were added to a stirred solution of mono-tert-butyl malonate (
Rf: 0.25 (n-hexane/EtOAc, 19:1).
IR (NaCl): 3087, 2980, 1748, 1732, 1650, 1145, 994 cm−1.
1H NMR (300 MHz, CDCl3): 1.47 (9H, s, t-Bu), 3.32 (2H, s, CH2), 4.65 (2H, dt, J = 5.7, 1.5 Hz, OCH2), 5.27 (1H, dq, J = 10.5, 1.2 Hz, C=CHH), 5.37 (1H, dq, J = 17.4, 1.5 Hz, C=CHH), 5.91 (1H, ddt, J = 10.5, 17.4, 6.0 Hz, CH=CH2).
13C NMR (100 MHz, CDCl3): 27.9 (CH3 × 3), 42.8 (CH2), 65.8 (CH2), 82.1 (C), 118.6 (CH2), 131.6 (CH), 165.6 (C=O), 166.6 (C=O).
MS (EI, 70 eV): m/z = 200 [M+].
HRMS-EI: m/z [M+] calcd for C10H16O4: 200.1049; found: 200.1054.
1-Allyl 3-(tert-Butyl) 2-Bromomalonate (7)
n-BuLi (1.6 M solution in n-hexane, 1.2 mL, 1.95 mmol) was slowly added to a stirred solution of N-isopropylcyclohexylamine (0.32 mL, 2.0 mmol) at −40°C. After stirring for 30 minutes at the same temperature,
The solution prepared above in tetrahydrofuran (THF) (5 mL) was added to a stirred solution of bromine (0.10 mL, 2.0 mmol) in THF (10 mL) at −78°C. After stirring for 2 hours at the same temperature, the reaction was quenched by the addition of 0.3 M HCl (30 mL). The resultant mixture was extracted with CH2Cl2 (30 mL, twice). The combined organic layers were washed with saturated aqueous NaHCO3 (75 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified via flash chromatography on silica gel (n-hexane/EtOAc, 39:1) to afford bromide
Rf: 0.3 (n-hexane/EtOAc, 19:1).
IR (NaCl): 3089, 2982, 1763, 1742, 1650, 1371, 1140 cm−1.
1H NMR (300 MHz, CDCl3): 1.49 (9H, s, t-Bu), 4.71 (2H, dt, J = 5.7, 1.2 Hz, CH2), 4.78 (1H, s, CHBr), 5.30 (1H, dq, J = 10.2, 1.2 Hz, C=CHH) 5.40 (1H, dq, J = 17.1, 1.5 Hz, C=CHH), 5.92 (1H, ddt, J = 10.2, 16.8, 6.0 Hz, CH=CH2).
13C NMR (100 MHz, CDCl3): 27.6 (CH3 × 3), 43.7 (CH), 67.3 (CH2), 84.4 (C), 119.4 (CH2), 130.8 (CH), 163.2 (C=O), 164.7 (C=O).
MS (EI, 70 eV): m/z = 280, 278 [M+].
HRMS-EI: m/z [M+] calcd for C10H15BrO4: 278.1054; found: 278.1048.
1-(tert-Butyl) 3-(2-Oxoethyl)-2-Bromomalonate (8)
Ozone was bubbled through a stirred solution of
Rf: 0.3 (n-hexane/EtOAc, 1:1).
IR (NaCl): 3492, 2981, 1739, 1731, 1302, 1257, 1140, 845 cm−1.
1H NMR (300 MHz, CDCl3): 1.52 (9H, s, t-Bu), 4.79 (2H, s, OCH2), 4.89 (1H, s, CHBr), 9.62 (1H, s, CHO).
13C NMR (100 MHz, CDCl3): 27.6 (CH3 × 3), 42.9 (CH), 70.0 (CH2), 85.0 (C), 162.9 (C=O), 164.6 (C=O), 194.1 (C=O).
MS (FAB, 10 kV): m/z = 283, 281 [M+H+].
HRMS-FAB: m/z [M+H+] calcd for C9H13BrO5: 281.0025; found: 281.0020.
3-(Methoxy(methyl)amino)-3-Oxopropanoic Acid (10)
Meldrum’s acid (
Rf: 0.25 (CHCl3/MeOH/AcOH, 95:5:3).
IR (NaCl): 3449, 1736, 1633, 1392, 1177 cm−1.
1H NMR (300 MHz, CDCl3): 3.28 (3H, s, NMe), 3.55 (2H, s, CH2), 3.75 (3H, s, OMe).
13C NMR (100 MHz, CDCl3): 32.1 (CH3), 36.7 (CH2), 61.5 (CH3), 168.9 (C=O), 169.2 (C=O).
MS (EI, 70 eV): m/z = 147 [M+].
HRMS-EI: m/z [M+] calcd for C5H9NO4: 147.0532; found: 147.0531.
Allyl 3-(Methoxy(methyl)amino)-3-Oxopropanoate (11)
Cesium carbonate (5.13 g, 15.8 mmol) and allyl bromide (1.4 mL, 16.5 mmol) were added to a stirred solution of
Rf: 0.3 (n-hexane/EtOAc, 3:2).
IR (NaCl): 3087, 1742, 1670, 1249, 1160, 992 cm−1.
1H NMR (300 MHz, CDCl3): 3.22 (3H, s, NMe), 3.53 (2H, s, CH2), 3.71 (3H, s, OMe), 4.65 (2H, dt, J = 5.7, 1.5 Hz, OCH2), 5.25 (1H, dq, J = 10.5, 1.2 Hz, C=CHH), 5.35 (1H, dq, J = 17.1, 1.5 Hz, C=CHH), 5.93 (1H, ddt, J = 10.2, 16.8, 6.0 Hz, CH=CH2).
13C NMR (100 MHz, CDCl3): 32.1 (CH3), 40.0 (CH2), 61.3 (CH3), 65.8 (CH2), 118.6 (CH2), 131.6 (CH), 167.09 (C=O), 167.10 (C=O).
MS (EI, 70 eV): m/z = 187 [M+].
HRMS-EI: m/z [M+] calcd for C8H13NO4: 187.0845; found: 187.0844.
Allyl 2-Bromo-3-(Methoxy(methyl)amino)-3-Oxopropanoate (12)
n-BuLi (1.6 M solution in n-hexane, 1.7 mL, 2.7 mmol) was slowly added to a stirred solution of N-isopropylcyclohexylamine (0.45 mL, 2.7 mmol) at −40°C. After stirring for 30 minutes at the same temperature,
The solution prepared above in THF (5 mL) was added to a stirred solution of bromine (0.15 mL, 2.9 mmol) in THF (5 mL) at −78°C. After stirring for 1.5 hours at the same temperature, the reaction was quenched by the addition of 0.3 M HCl (30 mL). The resultant mixture was extracted with CH2Cl2 (30 mL, three times). The combined organic layers were washed with saturated aqueous NaHCO3 (75 mL), dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified via flash chromatography on silica gel (n-hexane/EtOAc, 7:3). The combined fractions containing
Rf: 0.4 (n-hexane/EtOAc, 3:2).
IR (NaCl): 3087, 2943, 1764, 1744, 1673, 1151, 989 cm−1.
1H NMR (300 MHz, CDCl3): 3.26 (3H, s, NMe), 3.77 (3H, s, OMe), 4.68 (1H, ddt, J = 6.0, 12.9, 1.5 Hz, CHH), 4.73 (1H, ddt, J = 5.7, 12.9, 1.5 Hz, CHH), 5.26 (1H, s, CHBr), 5.28 (1H, dq, J = 10.5, 1.5 Hz, C=CHH), 5.38 (1H, dq, J = 17.1, 1.5 Hz, C=CHH), 5.92 (1H, ddt, J = 10.5, 17.1, 5.7 Hz, CH=CH2).
13C NMR (100 MHz, CDCl3): 33.0 (CH3), 42.6 (CH), 61.5 (CH3), 67.3 (CH2), 119.3 (CH2), 131.0 (CH), 164.5 (C=O), 166.6 (C=O).
MS (EI, 70 eV): m/z = 267, 265 [M+].
HRMS-EI: m/z [M+] calcd for C8H12BrNO4: 264.9950; found: 264.9944.
2-Oxoethyl 2-Bromo-3-(Methoxy(methyl)amino)-3-Oxopropanoate (13)
Ozone was bubbled through a stirred solution of
Rf: 0.3 (n-hexane/EtOAc, 1:4).
IR (NaCl): 3417, 2985, 1766, 1747, 1667, 1155, 994 cm−1.
1H NMR (300 MHz, CDCl3): 3.27 (3H, s, NMe), 3.81 (3H, s, OMe), 4.74 (1H, d, J = 17.1 Hz, CHH), 4.81 (1H, d, J = 17.1 Hz, CHH), 5.38 (1H, s, CHBr), 9.62 (1H, s, CHO).
13C NMR (100 MHz, CDCl3): 32.9 (CH3), 41.5 (CH), 61.7 (CH3), 70.0 (CH2), 163.7 (C=O), 164.4 (C=O), 194.6 (C=O).
MS (EI, 70 eV): m/z = 269, 267 [M+].
HRMS-EI: m/z [M+] calcd for C7H10BrNO5: 266.9742; found: 266.9747.
General Procedure for the Intramolecular Darzens Reaction
A base (1.3 equiv) and an organocatalyst (0.1 equiv) were added to a stirred solution of the substrate (1 equiv) in the solvent (c = 0.1 M), and the resultant mixture was stirred at room temperature. After full consumption of the starting material, the reaction was quenched with saturated aqueous NH4Cl, and the resultant mixture was extracted with EtOAc. The combined organic layers were washed with brine, dried over Na2SO4, filtered, and concentrated in vacuo. The crude residue was purified via flash chromatography on silica gel (n-hexane/EtOAc) to afford the desired epoxide.
N-Methoxy-N-Methyl-2-Oxo-3,6-Dioxabicyclo[3.1.0]Hexane-1-Carboxamide (15)
Rf: 0.5 (CHCl3/MeOH, 19:1).
IR (NaCl): 2918, 1786, 1685, 1462, 1246, 1064 cm−1.
1H NMR (400 MHz, CDCl3): 3.29 (3H, s, NMe), 3.75 (3H, s, OMe), 4.37 (1H, br s, CH), 4.38 (1H, dd, J = 1.2, 11.2 Hz, CHH), 4.50 (1H, d, J = 11.2 Hz, CHH).
13C NMR (100 MHz, CDCl3): 32.2 (CH3), 60.0 (CH), 61.3 (CH3), 67.4 (CH2), 77.2 (C), 161.3 (C=O), 167.4 (C=O).
MS (EI, 70 eV): m/z = 187 [M+].
HRMS-EI: m/z [M+] calcd for C7H9NO5: 187.0481; found: 187.0480.
Chiral HPLC analysis (CHRALPAK IC, EtOH:n-hexane = 50/50, flow rate = 1.0 mL/min, λ = 220 nm), t r (minor) = 6.9 minutes, t r (major) = 10.1 minutes.
Footnotes
Authors’ Note
This paper is dedicated to Professor Chiaki Kuroda on the occasion of his 65th birthday.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by JSPS KAKENHI Grant number 17K08227 and partially supported by a grant from the Dementia Drug Resource Development Center, Project S1511016, the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.