Wieland–Miescher ketone (1)1,2 and its congeners (2 and 3)2,3 are classic compounds, but very versatile synthetic building blocks especially for terpenoids, ever since the use of (S)-proline (4) as a chiral catalyst for their asymmetric syntheses (Figure 1).4,5 It is worthy of note that the intramolecular aldol reaction was extended to enantioselective intermolecular aldol reactions, which opened the door of the chemistry of organomolecular catalysts flourishing in these days. Synthetic studies starting from these compounds 1-3 as well as the development of a synthetic protocol to synthesize Wieland–Miescher ketone (1) are still continuing, which means continuing importance of these compounds as chiral building blocks.
Wieland–Miescher ketone (1) and its derivative (2) and Hajos–Parish–Eder–Sauer–Wiechert ketone (3).
Bradshaw and Bonjoch described well on the various aspects relating to Wieland–Miescher ketone (1) in their review in 2012.6 In this review, some representative synthetic studies of higher terpenoids since 2012 starting from Wieland–Miescher ketone (1) and its analog (2) were covered.
The traditional protocol for the preparation of chiral Wieland–Miescher ketone (1) was not an easy task different from its congeners 2 and 3. When easily available (S)-proline (4) was used as a chiral catalyst in dimethylsulfoxide, the proline (4) decomposed partially due to longer reaction time and resulting crystals of the Wieland–Miescher ketone (1) had color. Removal of the color from the crystals was difficult by repeated recrystallization. Enantioselectivity of the reaction was moderate and enantioenrichment by fractional recrystallization was difficult because racemate crystallized preferentially. Lower solubility of (S)-proline (4) was also problematic. These drawbacks stimulated the development of new catalysts and protocols due to the importance of Wieland–Miescher ketone (1).7
Diverse proline amides as organomolecular catalysts were devised (Figure 2), in which a variety of anchors were placed on a proline motif to fix enamine intermediates 16 and 17 enantiomerically (Scheme 1). Higher solubility of these proline amides in organic solvents enabled increase of the activity of these catalysts to enhance reaction rate, improve enantioselectivity, and to diminish the amount of catalysts. Some of them are suitable for solvent-free aldol processes with a lower catalyst loading and some others are recyclable by immobilizing on solid support.
Some representative prolineamide organocatalysts for the syntheses of optically active Wieland–Miescher ketone (1).8-19
Catalytic cycle of asymmetric synthesis of Wieland–Miescher ketone (1) catalyzed by prolineamide organocatalysts.
Organocatalysts from other unnatural chiral sources for the syntheses of Wieland–Miescher ketone (1) were illustrated in Figure 3. Selection of an optimum catalyst from these organomolecular catalysts depends on its catalytic activity, availability of a chiral source, ease and number of steps for its preparation, and ease of synthetic operation to prepare Wieland–Miescher ketone (1).
Organocatalysts from other chiral sources for the syntheses of Wieland–Miescher ketone (1).16-26
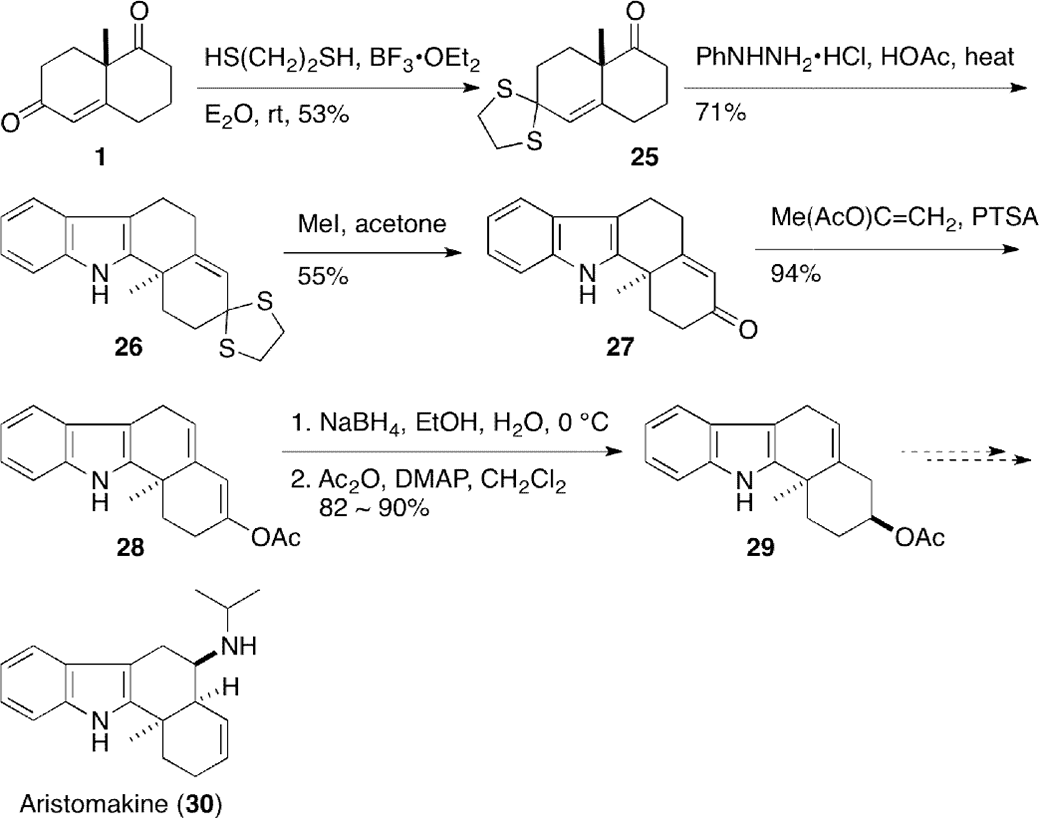
A synthetic approach toward (±)-aristomakine (30), an unusual alkaloid isolated from Aristotelia serrata, was reported by Perini and Gribble27 (Scheme 2). Thioacetalization of Wieland–Miescher ketone (1) proceeded without deconjugation of the double bond to give thioacetal 25, which was subjected to Fischer indole synthesis leading to indole 26. Attempts to furnish (±)-aristomakine (30) from acetate (29) were not fruitful.
Synthetic study of (±)-aristomakine (30).
The 3,4-seco-podocarpane trinorditerpenoids moluccanic acid methyl ester (37) was isolated from plant extracts of Aleurites moluccana and displays moderate cytotoxic activity against the HepG2 cell line with a half maximal inhibitory concentration (IC50 ) value of 32 µM, which was synthesized by Maier et al (Scheme 3)28 in racemic form. Starting from known decalone 31,29 introduction of a formyl group at C-8 followed by Robinson annulation provided tricyclic enone 32. Treatment with excess amount of Copper(II) bromide promoted the aromatization of the enone 32 in high yield, which was followed by deprotection and Dess–Martin periodinane (DMP) oxidation to furnish ketone 35. Regioselective Baeyer–Villiger oxidation and subsequent lactone ring opening furnished the target compound 37 in 35% yield from Wieland–Miescher ketone (1) in 14 linear steps.
Total synthesis of (±)-moluccanic acid methyl ester (37).
Giannis et al envisioned a synthetic study of C-nor-D-homo-steroid (45) based on Lewis acid-mediated Nazarov cyclization as a key step (Scheme 4).30 After addition of cyanohydrin trimethylsilyl-ether 40 to known ketone 39,31 deprotection and dehydration led to aryl-enone 42. Among a variety of Lewis acids, treatment with excess amount of titanium tetrachloride was effective to promote Nazarov cyclization leading to tetracyclic compound 43 as a single diastereomer. Deconjugation of the double bond of the enone and subsequent sodium borohydride reduction provided diol 44 as a single diastereomer. Birch reduction of the aryl moiety followed by conjugation of the double bond provided C-nor-D-homo-steroid core 45.
Synthetic study of C-nor-D-homo-steroid (45).
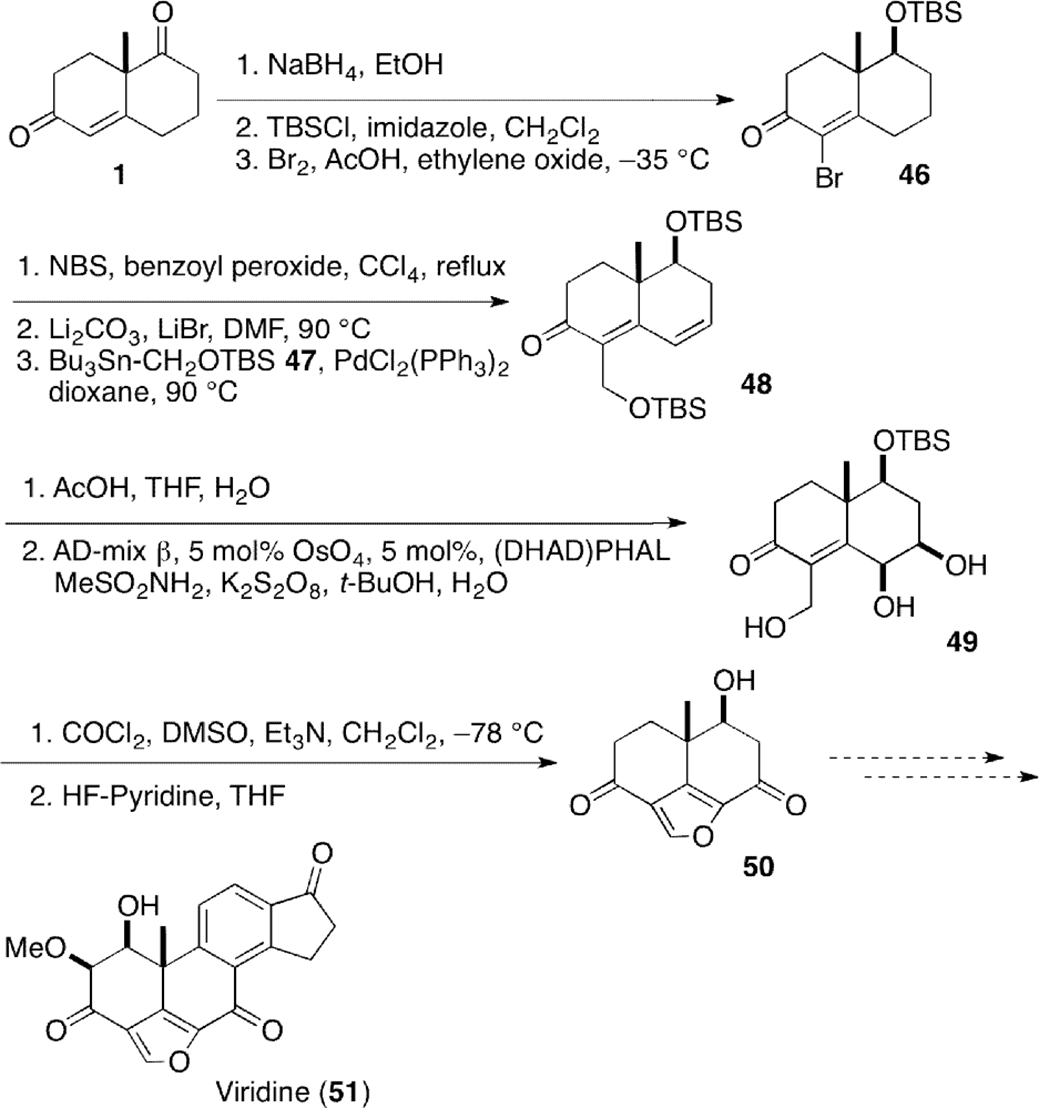
Synthesis of a truncated analog 50 of furanosteroid viridin (51), known as a potent inhibitor of the lipid kinase PI-3K, was carried out by Wright et al (Scheme 5).32 Reduction of Wieland–Miescher ketone (1) followed by protection as tert-butyldimethylsilyl (TBS) ether and subsequent bromination provided bromide 46. Subsequent radical bromination with n-bromosuccinimide at C-6 followed by dehydrobromination provided 4-bromo-4,6-dienone, which was introduced a formyl equivalent by palladium catalyzed coupling with an oxygenated stannane 4733 to give bis-silylether 48. Dihydroxylation of the dienone 48 proceeded regioselectively and stereoselectively from β-face of the molecule to furnish triol 49. Diacylfuran moiety was completed by Swern oxidation and subsequent cyclization to give diketone 50.
Synthetic study of viridin building block (50).
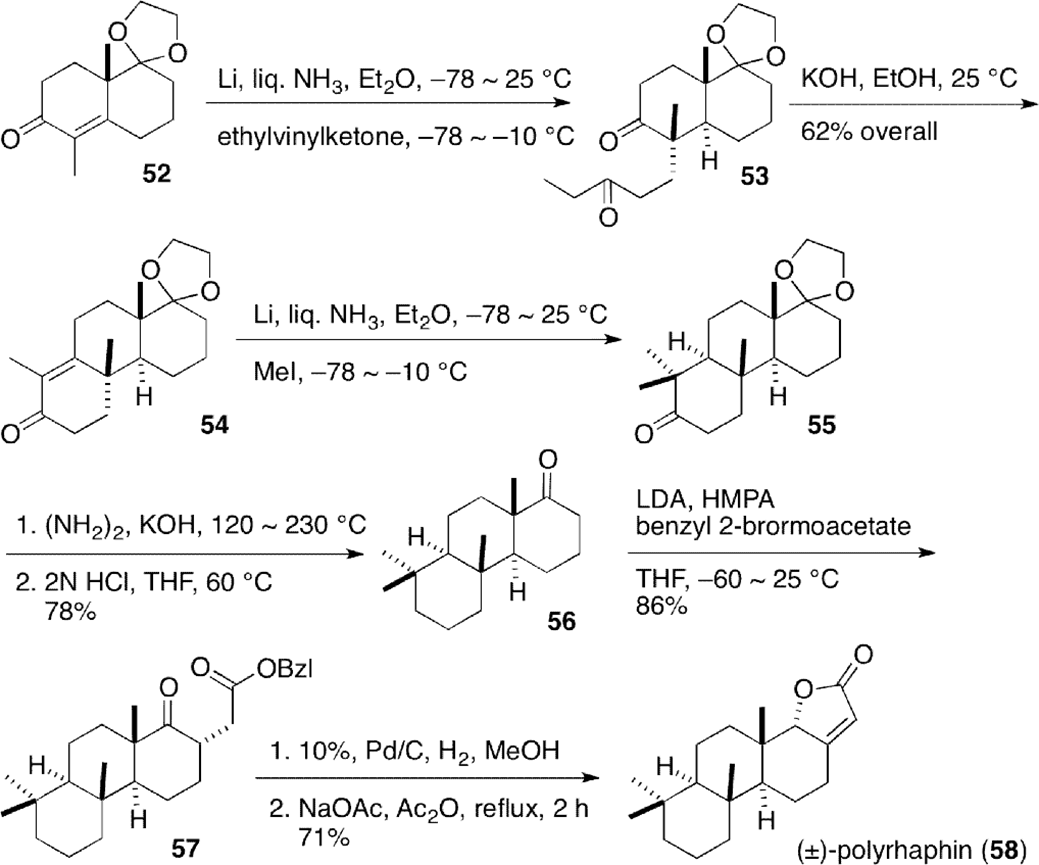
Total synthesis of spongian diterpenoid polyrhaphin D (58), a minor constituent of skin extracts of nudibranch Cadlina luteomarginata, was achieved by Xie et al (Scheme 6).34 Michael’s reaction of the enolate generated by lithium-ammonia reduction of known racemic enone 523 with penten-3-one proceeded from the α-face of the molecule to give trans-decalone 53, which was transformed into tricyclic-enone 54 by intramolecular aldol condensation. Reductive methylation with lithium in liquid ammonia and Wolff–Kishner reduction gave ketone 56, in which butenolide moiety was installed by dehydrative cyclization to furnish racemic polyrhaphin D (58).
Total synthesis of isospongian diterpenoid (±)-polyrhaphin D (58).
Echinoside A (67) is a potent antitumor triterpene saponin isolated from the sea cucumber Actinopyga echinites (JAEGER). Yu and Yu envisioned the synthesis of the ABC-fused ring skeleton of the aglycon of echinoside A (67) (Scheme 7).35 Enantiomerically enriched Wieland–Miescher ketone (1) was prepared according to the procedure reported by Bonjoch et al18,36 employing N-tosyl-(S)-binam-prolinamide (13) as an organocatalyst through a solvent-free asymmetric intramolecular aldol condensation of triketone 15. Thiomethylene unit at C-4 was introduced to enone 59 by Kirk–Petrow reaction37 to give sulfide 60. Reductive methylation of the sulfide 60 was effected with lithium in liquid ammonia to give trans-decalone 61. Reduction followed by protecting group manipulations led to MOM-ether 62, which was introduced a formyl group at C-8. Michael addition to buten-3-one and subsequent intramolecular aldol condensation led to enone 63, which was reduced with lithium in liquid ammonia to result in a single diastereomer, while catalytic hydrogenation gave a mixture of 2 diastereomers. Carbomethoxylation at C-13 provided ester 64, in which the double bond at C-13 was introduced by phenylselenylation and oxidation to afford enone 65. Conjugate addition of a methyl group at C-14 of the enone 64 followed by introduction of unsaturation furnished tricyclic precursor 66 of echinoside A (67).
Synthesis of the ABC skeleton 66 of the aglycon of echinoside A (66).
Inoue et al envisioned a convergent synthetic approach toward neurotoxic steroid batrachotoxin (88) isolated from the skins of Columbian poison-arrow frogs, by employing bridgehead radical coupling and Pd/Ni-promoted Ullmann reaction as key steps (Scheme 8).38
Synthetic study of the tetracyclic structure of batrachotoxin (88).
Synthesis of a coupling partner 73 for AB ring was carried out starting from known vinyl bromide 6839 (Scheme 8). Addition of lithiumvinyl ether proceeded from the convex face to give alcohol 69 as a single diastereomer, which was followed by acid catalyzed acetalization to provide acetal 70. A mixture of vinyl ether 70 was oxidized by ruthenium(III) chloride to afford carboxylic acid ester 70, which was treated with lithium iodide in pyridine to give carboxylic acid 72. After transformation to mesyl ester, the OMs group was replaced with phenyltelluride to acyl telluride 73.
Synthesis of a coupling partner for CD ring was started from diketone 74 (Scheme 9). Introduction of acetoxymethylene unit led to acetate 75 and subsequent transfer hydrogenation with Noyori catalyst 76 converted the acetate 75 into hydroxyketone 77 in 96% ee, which was protected as TBS ether 78. Enoltriflate 80 was obtained by the reaction with Commins reagent 79. Cleavage of the terminal double bond with OsO4 followed by Knoevenagel condensation of resulting aldehyde with malononitrile provided CD ring coupling partner 81.
Synthetic study of the tetracyclic structure of batrachotoxin (88).
Bridgehead radical coupling reaction of the two coupling partners 73 and 81 was effected with excess triethylborane under oxygen atmosphere to provide coupling product 83 as a diastereomeric mixture at C-11 (Scheme 10). The oxy radical 82 added to electron-deficient alkylidenemalonate moiety in 1,4-conjugate manner in preference to vinylbromide or enoltriflate moiety. Oxidation of the malononitrile group with magnesium monoperoxyphthalate 84 followed by hydrolysis provided ethyl ester 86. Although difficulty to connect C-8 and C-14 was anticipated due to their neopentyl positions, the intramolecular Ullmann coupling of 86 was successfully realized employing modified multimetallic Pd/Ni catalytic conditions by Weix et al40to furnish C ring cyclization product 87. It is worthy of note that all functional groups remained intact under the reaction conditions.
Synthetic study of the tetracyclic structure of batrachotoxin (88).
Taxol (109) has long been stimulating the interests of synthetic organic chemists because of its fascinating complex structure and intriguing physiological activity. Prunet et al described the synthesis of an advanced intermediate of taxol 108 via ring closing domino diene-yne metathesis of precursor 98 as a key step (Scheme 11).41 A synthesis of the precursor 98 started from TES-ether 89, which was cleaved by ozonolysis and subsequent thioesterification to give thioester 90 (Scheme 11). The thioester 90 was reduced with triethylsilane in the presence of palladium carbon, which was followed by acidic treatment to give hemiacetal 91 as a 1:1 epimeric mixture. After methylation, the resulting acetal 92 was transformed into hydrazone 93.
Synthetic study of an advanced intermediate of taxol (109).
As the coupling partner for the Shapiro reaction, racemic aldehyde 94, was chosen for easy availability (Scheme 12).42 Propargylation of ethyl isobutyrate 99 and subsequent hydrolysis provided acetylenic acid 100. After isomerization of the triple bond into internal position with t-BuOK, the resulting acid was transformed into Weinreb amide 101. Prenylation followed by cyanohydrin formation and reduction provided racemic aldehyde 94.
Synthetic study of an advanced intermediate of taxol (109).
Assemble by Shapiro reaction of the hydrazone 93 with the racemic aldehyde 94 was effected diastreoselectively to give diols 95 and 96 (Scheme 11). The diol 96 was protected as a carbonate, which was followed by hydrolysis of the acetal and subsequent reduction to furnish diol 97. Application of Grieco protocol led to requisite diene-yne 98.
With the diene-yne 98 in hand, the key diene-yne metathesis reaction was investigated, in which Zhan-1b precatalyst 103 was optimum due to its availability to realize the metathesis furnishing desired tricyclic core 105 of taxol (109) along with diene metathesis product 104 as a major product (Scheme 13). An equimolar mixture of diene-yne 98 and the diene metathesis product 104 was again submitted to the metathesis reaction in the presence of an equimolar amount of Zhan-1b precatalyst 103, which presented the tricyclic core 105 in 47% yield. Epoxidation of the tricyclic core 105 with excess meta-chloroperoxybenzoic acid (MCPBA) proceeded initially at bridgehead position from the upper face to give selectively trans-1,3-bisepoxide 106, which was treated with phenyllithium to provide benzoate 107. Radical ring opening of the bis-epoxide ring of the benzoate 107 mediated by Ti(III) furnished triol 108, an advanced intermediate for taxol (109) synthesis.
Synthetic study of an advanced intermediate of taxol (109).
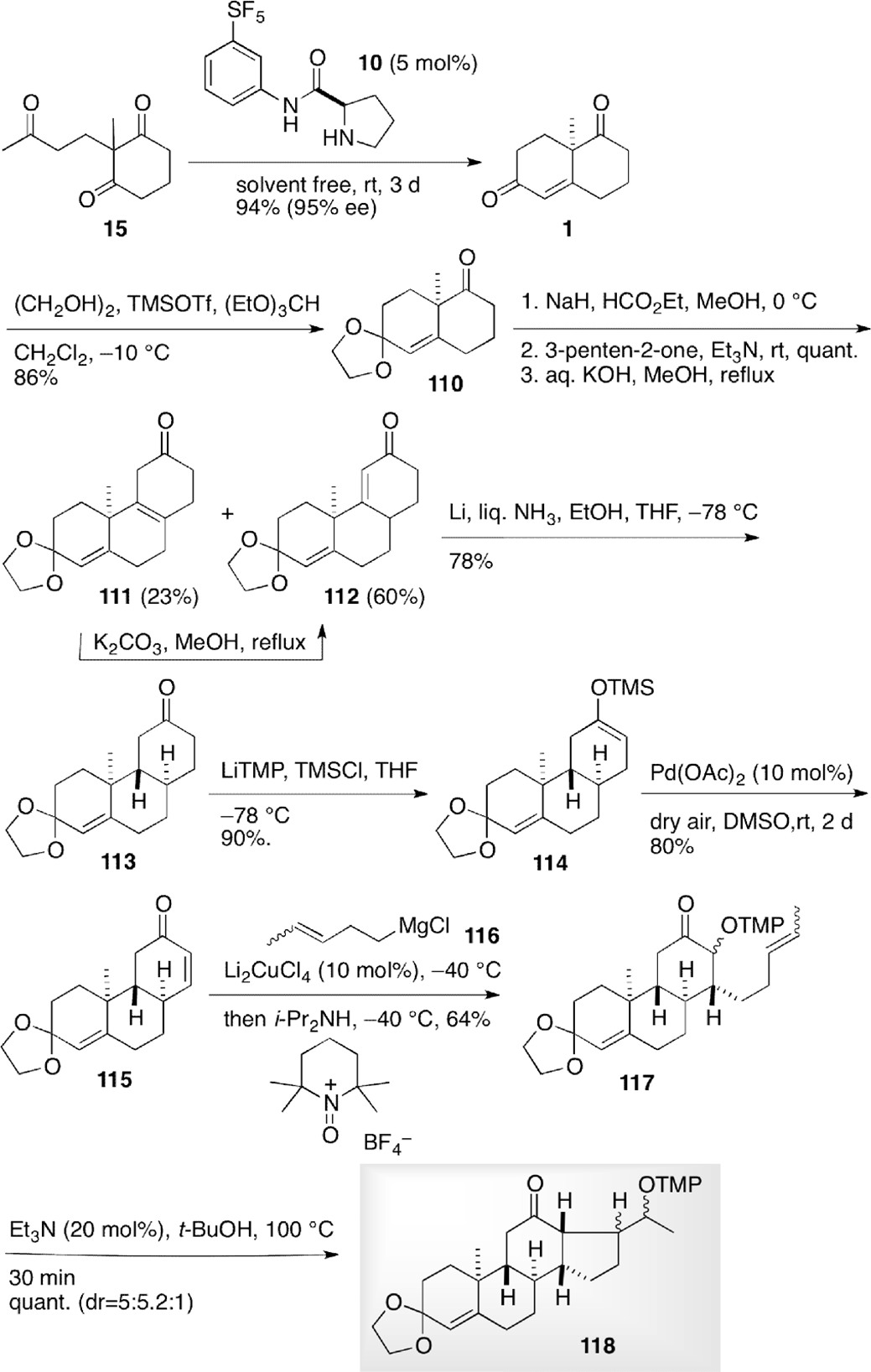
Jahn et al envisioned total synthesis of ent-pregnanolone sulfate (126) in order to shed light on the action and binding mode of inhibitory steroids on the N-methyl-D-aspartate receptors, which was accomplished by employing radical reaction to form D-ring as a key step (Scheme 14).15 The asymmetric intramolecular aldol condensation of triketone 15 was carried out with organocatalyst 10 to provide Wieland–Miescher ketone (1). The catalyst 10 was the most efficient among other catalysts in high enantioselsectivity under solvent-free conditions in shorter reaction time. Toward the total synthesis of the target compound 126, regioselective protection of the carbonyl group at C-3 was required, which was enabled without migration of the double bond at C-4 with ethyleneglycol and trimethylsilyl triflate as an initiator to give acetal 110. Introduction of a formyl group at C-8 followed by Michael’s reaction and aldol condensation delivered tricyclic compounds 111 and 112. Reduction of the enone 112 with lithium in liquid ammonia gave trans-BC tricyclic compound 113. Transformation to silylenolether 114 with lithium tetramethylpiperidide followed by Saegusa oxidation led to enone 115. Conjugate addition of Grignard reagent 116 and subsequent trap of resulting enolate with tetramethylpiperidine N-oxide gave precursor for the radical reaction 117. The key radical reaction was performed at 100°C to furnish tetracyclic compound 118 as a mixture of 3 diastereomers.
Total synthesis of ent-pregnanolone sulfate (126).
Methylation of 118 gave an inseparable mixture of diastereomers 119 (Scheme 15), which was transformed into the enol nonaflate 121. The alkoxyamino group of 121 was oxidized with MCPBA to result in methylketone 122, which was subjected to catalytic hydrogenation to provide ketone 123. This protocol solved the difficulty to remove the carbonyl group at C-12. Presence of solid supported Amberlite base was crucial in the hydrogenation. After hydrolysis of the aectal 123, catalytic hydrogenation in the presence of 4-methoxypyridine proceeded from the upper face of the enone to provide cis-AB ring structure. Regioselective sodium borohydride reduction followed by sulfation completed the synthesis of ent-pregnanolone sulfate (126)
Total synthesis of ent-pregnanolone sulfate (126).
Gomphostenin (137), isolated from Gomphostemma niveum, exhibits antimalarial properties. Synthetic approach toward gomphostenin (137) from known decalone 12743,44 was shown by Yadav et al45 (Scheme 16).
Synthetic study of antimalarial gomphostenin (137).
Cleavage of the double bond of 127 by ozonolysis and subsequent reduction provided diol 128. Ketone 129 was obtained by selective protection of the primary hydroxyl group followed by DMP oxidation. Hydroboration of olefin 130 derived by Wittig methylenation of 129 proceeded non-stereoselectively to give primary alcohol 131 and 132. Deprotection of the acetal of α-alcohol 131, which was followed by protection of the primary alcohol and subsequent Saegusa oxidation furnished enone 133. Addition of mehyllithium, deprotection of the benzylether, and DMP oxidation provided aldehyde 134. Wittig olefination with ylide 135 followed by oxidative rearrangement with pyridinium chlorochromate (PCC) led to enone 136. Attempts of isomerization of the exo-double bond leading to gomphostenin (137) were not successful, resulting in a mixture of products, recovery of starting material, and formation of unknown products.
Dysideanone B (142), isolated from the South China Sea sponge Dysidea avara, possesses an unprecedented carbotetracyclic skeleton and potential anticancer property. Its core structure 141 was prepared by Haque et al46 from known decalone 138,3,47 which was treated with arylmethylbromide and lithium metal under sonication to deliver alcohol 140 stereoselectively (Scheme 17). Lewis acid-mediated rearrangement provided tetracyclic product 141.
Synthetic study of dysideanone B (142).
3β-Hydroxy-7β-kemp-8(9)-en-6-one (165), a kempane diterpenoid which was isolated from the soldier defense secretion of the higher termites Nasutitermes octopolis and features an array of 8 stereogenic centers in a bowl-shaped structure, which was elaborated via domino diene-yne metathesis as a key step by Metz et al (Scheme 18).47
Total synthesis of 3β-hydroxy-7β-kemp-8(9)-en-6-one (165).
The carbonyl group at C-3 of Wieland–Miescher ketone (1) was regioselecively protected as a dithiolate without migration of the double bond at C-4 (Scheme 18). Monomethylation was effected with sodium enoxyborate to give a mixture of diastereomers, which were unified by potonation to ketone 143 having an equatorial methyl group. Addition of lithiated Wittig–Horner reagent followed by elimination with sodium hydride provided enol ether 144, which was hydrolyzed to give aldehyde 145. Homologation of the formyl group of aldehyde 145 was carried out by Wittig reaction and subsequent hydrolysis leading to aldehyde 146. Introduction of isopropenyl group followed by copper catalyzed hydrolysis of dithiolane furnished enone 147. Introduction of an equatorial methyl group at C-2 was carried out in the same manner via enoxyborate protocol leading to enone 148. Reduction of the enone with lithium in liquid ammonia provided trans-decalone, which was transformed into enone 150 through regioselective silylenol ether formation and subsequent Mukaiyama dehyderogenation with sulfinimidoyl chloride 149.48 Conjugate addition of propargylaluminate generated from propargylstanane 151 to the enone 150 proceeded on β-face of the molecule to avoid steric repulsion between the angular methyl group and to give ketone 152. An attempt to introduce an axial allyl group was carried out to an enolate generated by cleavage of silylenol ether of the ketone 152 to provide a mixture of C- and O-allylated products 153 and 154. Without separation, the mixture was heated to promote Claisen rearrangement of the allyl vinyl ether 154 and converged to the diene-yne 153 having complete diastereoselectivity.
Prior to the metathesis reaction, the carbonyl group of 153 was reduced with l-selectride to arrange the correct stereochemistry of alcohol 155 (Scheme 19). The key domino diene-yne metathesis of the alcohol 155 was managed successfully with Grubbs second-generation catalyst to furnish diene 156 having requisite tetracyclic core in high yield. Since the double bond in 7-membered ring was more active toward epoxidation, the double bond was saved as diol. Osmium tetroxide attacked from convex face of the bowl-shaped molecule 156 to give diol 157. Epoxidation of the olefin in 5-membered ring was effected with methyl(trifluoromethyl)dioxirane generated in situ to provide β-epoxide 158, which was transformed into thionocarbonate 159. Treatment with diazaphospholidine regenerated the double bond in 7-membered ring to provide vinylepoxide 161, which was sensitive enough to undergo rearrangement on silica gel to give β,γ-unsaturated ketone 162. Epimerization at C-7 was accomplished by conjugate addition of thiophenol to enone 163 and elimination of phenylsulfinic acid to culminate in the total synthesis of β-hydroxy-7β-kemp-8(9)-en-6-one (165).
Total synthesis of 3β-hydroxy-7β-kemp-8(9)-en-6-one (165).
(+)-Taondiol (176) is a pentacyclic meroterpenoid involving a diterpenoid moiety, which was isolated from algal metabolites from Stypopodium zonale, while its enantiomer (–)-taondiol was isolated from marine alga Taonia atomaria and exhibits lethargic behavior and narcosis at 10 µg/mL levels. Their absolute stereostructures were unknown.
Dethe et al illustrated the total synthesis of (+)-taondiol (176) through combining diterpene core 172 with hydroxyquinol 173 by Lewis acid-mediated Friede–Crafts reaction (Scheme 20).49
Total synthesis of (+)-taondiol (176).
After regioselective protection of the saturated ketone of Wieland–Miescher ketone derivative 2, thermodynamically controlled alkylation with 1-chloro-3-pentanone proceeded from the α-face of the substrate, which was followed by aldol condensation to provide tricyclic enone 157. Reductive methylation of the enolate generated with lithium in liquid ammonia gave ketone 168, whose carbonyl group was reduced and protected as benzyl ether 169. In order to prevent debenzylation, the double bond was reduced by transfer hydrogenation according to the method by Shenvi et al50 to give all-trans tricyclic core 170. α-Methylation followed by epimerization gave α-methyl ketone 171. α,β-Unsaturated aldehyde moiety was installed via the addition of chloromethyllithium according to the method of Nozaki-Yamamoto,51 and subsequent reduction provided alcohol 172. The Friedel–Crafts coupling with hydroxyquinol 173 with BF3•OEt2 effected pentacyclic compound 174. Conversion to triflate followed by palladium-catalyzed transfer reduction led to ether 175. Deprotection of the methyl ether and subsequently benzyl ether completed the total synthesis of (+)-taondiol (177) thereby establishing absolute stereostructure.
JBIR-03 (188), isolated at first from Dichotomomyces cejpii var. cejpii NBRC 103559 and from a marine-derived strain of Aspergillus oryzae and of D. cejpii, is a hexacyclic indole diterpenoid, which exhibits anti-MRSA, antifungal, and insecticidal activities without cytotoxicity. Asporyzin C (187), a pentacyclic indole diterpenoid isolated together with JBIR-03 (188) from A. oryzae, exerts potent antibacterial activity against Escherichia coli. These compounds were elaborated Pd(II)-mediated indole ring formation as a key step by Kuwahara et al for the first time (Scheme 21).52
Total synthesis of JBIR-03 (188) and asporyzin C (187).
After the protection of known hydroxy-ketone 177a53as TBS ether, alkylation of 177b with bromide 178 followed by hydrolysis of the enol ether provided phosphonate 179 as an epimeric mixture. Intramolecular Horner–Wadsworth–Emmons reaction led to cyclopentenone 180 as a single diastereomer. Reduction with l-selectride gave stereoselectively allylic α-alcohol, whose Simmons–Smith cyclopropanation was directed by chelation control of the hydroxyl group to provide cyclopropyl alcohol. The Parikh–Doering oxidation of the resulting cyclopropyl alcohol afforded cyclopropylketone 181. Reductive cleavage of the cyclopropane ring was achieved with sodium naphthalenide to prepare α-methyl group at C-3 and the resulting enolate was trapped with Comins’ reagent 79 to give enol triflate 182, which was coupled with stannane 183 to afford precursor 184. The indole moiety was effected by Pd(II)-mediated oxidative ring formation to furnish pentacyclic compound 185. After deprotection of the TBS group, side chain was installed by cross metathesis with 2-methyl-3-buten-2-ol to give diol 186. Deprotection of the BOC group at reduced pressure completed the total synthesis of asporizyn C (187). Subsequent palladium catalyzed cyclization proceeded stereoselectively to furnish JBIR-03 (188).
(+)-Decaturin C (201), a constituent of Penicillium thiersii, is a diterpenoid coupled with spiro-linkage with nicotinate-containing polylketide and exhibits an anti-insectant activity. The first total synthesis was achieved via spiro-cyclization as a key step by Takikawa et al (Scheme 22).54
Total synthesis of decaturin C (201).
Known enone 189 was reduced with lithium in liquid ammonia and resulting intermediary enolate was trapped with Manders’ regent to provide ester 190. Conjugate addition to 2-penten-3-one proceeded from the α-face of 190 and successive intramolecular aldol condensation provided tricyclic enone 191 as a single diastreomer. Reduction with lithium aluminum hydride (LAH) led to diol 192. Selective oxidation of the allyl alcohol and subsequent protection of remaining primary alcohol provided enone 193. Reductive methylation with lithium in liquid ammonia under high dilution conditions gave all trans-tricyclic compound 194 in reproducible yield. Deprotection of the TBS ether followed by treatment with trimethyl orthoformate furnished ketal 195. After oxidation with pyridinium dichromate, a methyl group was introduced to give ketone 196, which was led to enone 197 by phenylselenylation and selenoxide elimination. Addition of metyllithium proceeded from the α-face of the enone 197 selectively to give allylalcohol, which was epoxidized with MCPBA to provide α-epoxy alcohol 198. Steric congestion around the hydroxyl group might disturb chelation of the hydroxyl group to allow reaction of MCPBA from less hindered α-face. Dehydration of the alcohol 198 led to exo-olefin 199. The key spirocyclization was carried out with pyrone 200 under refluxing conditions in anisole. Hydrolysis of the ketal completed the total synthesis of decaturin C (201).
The jujubosides are saponin natural products, which are reported to have immunoadjuvant, anticancer, antibacterial, antifungal, and antisweet activities. The triterpene component, jujubogenin (216) contains a unique tricyclic ketal motif comprising the DEF ring system. A synthetic approach toward the total synthesis of jujubogenin (216) was described by Karimov et al employing a sterically demanding intermolecular Diels–Alder reaction to assemble the C-ring and a tandem Wolff rearrangement-intramolecular ketene hetero-Diels–Alder reaction to form the DF-ring system. Acid catalyzed cyclization of the resulting bicyclic enol ether then closed the E-ring to provide the hexacyclic core of jujubogenin (216) (Scheme 23).55
Synthesis of the hexacyclic triterpene core of the jujuboside saponins (216).
Diene 203 was obtained by the addition of vinylmagnesium bromide followed by dehydration starting from ketone 202, which was prepared from Wieland–Miescher ketone (1) in 7 steps (Scheme 23). The Diels–Alder reaction was carried out without Lewis acid to give adducts 205 and 204 in a 2:1 ratio. After methanolysis of the major anhydride 205 to tri-ester, catalytic hydrogenation with Crabtree catalyst proceeded from the α-face of the molecule to afford all trans tri-ester 207, whose ester groups were transformed separately one by one in subsequent steps. Less hindered ester at C-13 was regioselectively reduced and oxidized to provide aldehyde 208. Wittig methylenation of the aldehyde 208 and subsequent reduction of 2 esters afforded diol 209. 2-Iodoxybenzoic acid (IBX) oxidation under sonication resulted in selective oxidation of an equatorial hydroxymethylene group to afford equatorial aldehyde 210, while an axial hydroxymethylene group was oxidized by Dess–Martin reaction. Protection of the remaining alcohol as isopropyldiethylsilyl ether was optimum due to stability and cleavage in the later stage. The formyl group was transformed into methylketone 211 by the addition of methyllithium and subsequent Dess–Martin oxidation. Ozonolysis of the olefin 211 followed by Horner–Wadsworth–Emmons olefination of resulting aldehyde installed side chain to give triene 213 as a 2:1 mixture of Z- and E-enol ethers. Introduction of a formyl group and subsequent diazotransfer reaction afforded diazoketone 214. The key tandem Wolff rearrangement-intramolecular ketene hetero-Diels–Alder reaction of the diazoketone 214 was managed by irradiation at 310 nm in C6D6 to deliver the desired pentacyclic bis(enolether) 215 having requisite functionality for further transformations, while silver-mediated reaction did not proceed without irradiation. However, the cycloadduct 215 readily decomposed upon purification on silica gel or alumina. The relative stereochemistry at C23 was not assigned due to its instability.
Toward this end, diazoketone 217 having benzylether in the side chain was prepared according to the same protocol (Scheme 24). The key tandem Wolff rearrangement-intramolecular ketene hetero-Diels–Alder reaction provided stable pentacyclic compound 218, whose stereochemistry at C-23 was opposite to that of jujubosides. Deprotection of silylether followed by treatment with dichloroacetic acid gave desired hexacyclic keto-ketal 219. Addition of methyllithium and subsequent catalytic hydrogenation led to diol 220. Treatment with Martin’s sulfurane 221 allowed regioselective dehydration of the side chain alcohol to afford isomeric olefins 222 and 223.
Synthesis of the hexacyclic triterpene core of the jujuboside saponins (222).
Schiglautone A (242), isolated from Schisandra glaucescens, is a triterpenoid having unprecedented 6/7/9 fused tricyclic skeleton and displays weak cytotoxic activity against HeLa, HepG2, and SGC-7901 cancer cell lines. A platform to construct central core of schiglautone A (242) was exploited by Le Chapelain by coupling aldehyde 229 and ketone 237 (Scheme 25).56
Synthetic study of schiglautone A (242).
Introduction of a methyl group to an α-position of known ketone 224 and subsequent α-oxygenation using molecular oxygen in the presence of triethylphosphite provided hydroxy-ketone 225 (Scheme 25). Reduction followed by cleavage of resulting diol with Pb(OAc)4 led to keto-aldehyde 226. An acetylenic moiety was introduced by Seyferth–Gilbert homologation to give alkyne 227. Baeyer–Villiger oxidation afforded acetate 228, which was hydrolyzed and oxidized to furnish the coupling partner aldehyde 229.
Synthesis of another coupling partner, ketone 237, started from 3-methoxyacetophenone 230 by enantioselective transfer hydrogenation employing Noyori’s catalyst to deliver secondary alcohol 231 with 98% ee. (Scheme 26).57 An introduction of a homoprenyl group to the alcohol 231 was implemented according to the protocol of Aggarwal et al.58 Treatment of carbazole derived from the alcohol 231 with s-butyllithium and boronate ester 232 and subsequent addition of magnesium bromide in methanol enabled 1,2-alkyl migration. Protodeboronation of the intermediary boronic ester was effected with TBAF•3H2O in toluene to afford substituted anisole 233 in good yield with a satisfactory enantiomeric excess of 90%. Reduction of the anisole 233 with lithium in liquid ammonia followed by hydrolysis provided enone 234. Due to modest diastereoselectivity of conjugate addition of AlMe3 in the presence of CuI to the enone 234, the methyl group was introduced by cleavage of cyclopropane ring. Thus, Corey–Bakshi–Shibata reduction of the enone 234 led to allylic alcohol 235, in which major diastereomer was subjected to cyclopropanation to give cyclopropyl alcohol 236. PCC oxidation followed by reduction with lithium in liquid ammonia afforded an additional coupling partner 237.
Synthetic study of schiglautone A (242).
Coupling of aldehyde 229 and cyclohexanone 237 was conducted by aldol reaction with lithium diisopropylamide (LDA) and subsequent trifluoroacetylation followed by elimination to furnish enone 238 (Scheme 27). Conjugate reduction with copper (II) iodide/LAH delivered cyclohexanone 239. In order to introduce a methyl group regio- and stereoselectively, cyclopropanation of thermodynamically stable silylenol ether of the ketone 239 was attempted to give cyclopropyl ether 240, whose hydrolysis resulted in ring opening to isolate desired ketone 241 along with other stereoisomer and compounds having cyclopropyl side chain.
Synthetic study of schiglautone A (242).
An alternative synthetic approach toward schiglautone A (242) was described employing samarium(II) iodide (SmI2)-mediated pinacol coupling to construct 9-membered ring by Werner and Kalesse (Scheme 28).59
Synthetic study of schiglautone A precursor (252).
Ring expansion of known ketone 3160 with ethyl diazoacetate and BF3•OEt2 followed by decarboxylation of resulting ester furnished 7-membered ketone 243, which was led to enone 244 by Saegusa oxidation. 4-Pentenyl side chain was introduced by the conjugate addition of the Grignard reagent 245. Since trapping of the enolate generated by conjugated addition with trimethylsilyl chloride (TMSCl) was not possible, silylenol ether 246 was obtained separately with LDA and TMSCl. Treatment with sodium iodide and MCPBA gave an α-iodide,61 which delivered enone 247 by elimination with DBU. Saegusa oxidation of the enol ether 247 resulted in low yield. 1,4-Conjugate addition of methylmagnesium bromide proceeded from the less hindered convex face of the molecule to afford ketone 248 stereoselectively. Addition of vinylmagnesium bromide followed by oxidative rearrangement with PCC provided E-enal 249. OsO4 and subsequently sodium periodate oxidation gave dialdehyde 250. The key SmI2-mediated pinacol coupling successfully closed a 9-membered ring to furnish diol 251 as a diastereomeric ratio of 2:1. Regioselective protection of the allylic alcohol followed by IBX oxidation afforded plausible precursor 252 having requisite stereocenters for schiglautone A (252) synthesis.
Since its invention, Wieland–Miescher ketone (1) and its derivatives have been widely used as starting materials of natural product syntheses, because they equip already decalone framework with requisite functionalities for further transformations, such as regioselective reduction or protection of 2 carbonyl groups, reductive alkylation leading to trans-decalone, catalytic hydrogenation leading to cis-decalone, annulation or alkylation on both carbonyl groups, etc. Moreover, the development of enantioselective synthesis employing amino acids as chiral catalysts enhanced chiral Wieland–Miescher ketone (1) as a versatile chiral building. In addition, the enantioselective intramolecular aldol reaction triggered the chemistry of organomolecular catalysts, which have brought the prosperity of the chemistry of organomolecular catalysts in these decades. Although the original enantioselective synthesis of Wieland–Miescher ketone (1) employing (S)-proline as a chiral catalyst had some issues, the development of the chemistry of organomolecular catalysts allowed to supply various organomolecular catalysts specified for Wieland–Miescher ketone (1) synthesis. Improvement of availability of chiral Wieland–Miescher ketone (1) facilitated furthermore the synthetic studies of natural products starting from Wieland–Miescher ketone (1). In this article, some representative efforts in the synthetic studies of higher terpenoids starting from Wieland–Miescher ketone (1) and its derivative (2) as chiral building blocks have been reviewed since 2012, where diverse synthetic strategies have been exemplified to construct complex structural units involving diterpenoids, triterpenoids, a meroterpenoid, and steroids. Since mother nature has potential to produce many natural products having intriguing structures as well as physiological activities, efforts will continue to synthesize natural products, in which the important status of chiral Wieland–Miescher ketone (1) will be kept for the syntheses of higher terpenoids.62
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
ORCID iD
Hisahiro Hagiwara
References
1.
WielandP.MiescherK. Über die Herstellung mehrkerniger ketone. Helv Chim Acta. 1950;33(7):2215-2228. doi:10.1002/hlca.19500330730
2.
RamachandranS.NewmanMS. The title was inserted to be " 1,6-DIOXO-8a-METHYL-1,2,3,4,6,7,8,8a-OCTAHYDRONAPHTHALENE". Org Synth. 1961;41:38-41.
3.
HagiwaraH.UdaH. Optically pure (4aS)-(+)- or (4aR)-(-)-1,4a-dimethyl-4,4a,7,8-tetrahydronaphthalene-2,5(3H,6H)-dione and its use in the synthesis of an inhibitor of steroid biosynthesis. J Org Chem. 1988;53(10):2308-2311. doi:10.1021/jo00245a033
4.
EderU.SauerG.WiechertR. New type of asymmetric cyclization to optically active steroid CD partial structures. Angew Chem Int Ed Engl. 1971;10(7):496-497. doi:10.1002/anie.197104961
5.
HajosZG.ParrishDR. United States Patent US3975442; 1971.
6.
BradshawB.BonjochJ. The Wieland-Miescher ketone: a journey from Organocatalysis to natural product synthesis. Synlett. 2012;23(03):337-356. doi:10.1055/s-0031-1290107
SrivastavaV. PEG-Solvent system for L-proline catalyzed Wieland-Miescher ketone synthesis. Current Organocatal. 2014;1(1):2-6. doi:10.2174/2213337201666140226000527
10.
AlmaşiD.AlonsoDA.NájeraC. Prolinamides versus Prolinethioamides as Recyclable Catalysts in the Enantioselective Solvent-Free Inter- and Intramolecular Aldol Reactions. Adv Synth Catal. 2008;350(16):2467-2472. doi:10.1002/adsc.200800430
11.
PanL.DingX.DingJet al. Design and Synthesis of L-Proline Derivatives as Enantioselective Organocatalysts for Synthesis of the ( S )-Wieland-Miescher Ketone. ChemistrySelect. 2017;2(36):11999-12005. doi:10.1002/slct.201702075
12.
RubioOH.Fuentes de ArribaÁngel L..MonleónLMet al. Bifunctional organocatalysts based on a carbazole scaffold for the synthesis of the Hajos–Wiechert and Wieland–Miescher ketones. Tetrahedron. 2015;71(8):1297-1303. doi:10.1016/j.tet.2014.12.079
13.
ZhangX-M.WangM.Y-QTet al. Prolinamide/PPTS-Catalyzed Hajos-Parrish Annulation: efficient approach to the tricyclic core of Cylindricine-Type alkaloids. Synlett. 2008;18:2831-2835.
14.
Vizcaíno-MillaP.SansanoJM.NájeraC.FiserB.Gómez-BengoaE. Pyrimidine-Derived Prolinamides as recoverable bifunctional Organocatalysts for enantioselective inter- and intramolecular aldol reactions under solvent-free conditions. European J Org Chem. 2015;2015(12):2614-2621. doi:10.1002/ejoc.201500007
15.
KaprasV.VyklickyV.BudesinskyMet al. Total synthesis of ent-Pregnanolone sulfate and its biological investigation at the NMDA receptor. Org Lett. 2018;20(4):946-949. doi:10.1021/acs.orglett.7b03838http://www.ncbi.nlm.nih.gov/pubmed/29364682
16.
D’EliaV.ZwicknaglH.ReiserO. Short alpha/beta-peptides as catalysts for intra- and intermolecular aldol reactions. J Org Chem. 2008;73(8):3262-3265.doi:10.1021/jo800168hhttp://www.ncbi.nlm.nih.gov/pubmed/18341352
17.
Bañón-CaballeroA.GuillenaG.NájeraC.FaggiE.SebastiánRM.VallriberaA. Recoverable silica-gel supported binam-prolinamides as organocatalysts for the enantioselective solvent-free intra- and intermolecular aldol reaction. Tetrahedron. 2013;69(4):1307-1315.doi:10.1016/j.tet.2012.11.097
18.
BradshawB.Etxebarría-JardiG.BonjochJ.ViózquezSF.GuillenaG.NájeraC. Efficient solvent-free Robinson Annulation protocols for the highly enantioselective synthesis of the Wieland-Miescher ketone and analogues. Adv Synth Catal. 2009;351(14-15):2482-2490. doi:10.1002/adsc.200900321
19.
BradshawB.Etxebarria-JardíG.BonjochJ.ViózquezSF.GuillenaG.ofNS. S)-8a-Methyl-3,4,8,8a-Tetrahydro-1,6- (2H,7H)-Naphthalenedione via N-Tosyl-(Sa)-binam-L-prolineamide Organocatalysis. Org Synth. 2011;88:330-341.
20.
CañellasS.AyatsC.HenselerAH.PericàsMA. A highly active polymer-supported catalyst for asymmetric Robinson Annulations in continuous flow. ACS Catal. 2017;7(2):1383-1391. doi:10.1021/acscatal.6b03286
21.
ZhouP.ZhangL.LuoS.ChengJ-P. Asymmetric synthesis of Wieland-Miescher and Hajos-Parrish ketones catalyzed by an amino-acid-derived chiral primary amine. J Org Chem. 2012;77(5):2526-2530. doi:10.1021/jo202433vhttp://www.ncbi.nlm.nih.gov/pubmed/22316216
22.
DaviesSG.SheppardRL.SmithAD.ThomsonJE. Highly enantioselective Organocatalysis of the Hajos-Parrish-Eder-Sauer- Wiechert reaction by the β-amino acid cispentacin. Chem Commun. 2005;30:3802-3804.
MoriK.KatohT.SuzukiT.NojiT.YamanakaM.AkiyamaT. Chiral phosphoric acid catalyzed desymmetrization of meso-1,3-diones: asymmetric synthesis of chiral cyclohexenones. Angew Chem Int Ed Engl. 2009;48(51):9652-9654. doi:10.1002/anie.200905271http://www.ncbi.nlm.nih.gov/pubmed/19924752
25.
Fuentes de ArribaÁngel L.SeisdedosDG.SimónL.AlcázarV.RaposoC.MoránJR. Synthesis of monoacylated derivatives of 1,2- cyclohexanediamine. evaluation of their catalytic activity in the preparation of Wieland-Miescher ketone. J Org Chem. 2010;75(23):8303-8306. doi:10.1021/jo101723vhttp://www.ncbi.nlm.nih.gov/pubmed/21058658
26.
WangY.JägerA.GrunerM.LübkenT.MetzP. Enantioselective Total Synthesis of 3β-Hydroxy-7β-kemp-8(9)-en-6-one, a Diterpene Isolated from Higher Termites. Angew Chem Int Ed Engl. 2017;56(50):15861-15865.doi:10.1002/anie.201708561http://www.ncbi.nlm.nih.gov/pubmed/28960721
27.
PeriniRB.GribbleGW. A synthetic approach to (±)-aristomakine. A synthetic approach to (±)-aristomakine. ARKIVOC. 2018;part v:75-84.
28.
UshakovDB.RajaA.FrankeR.SasseF.MaierME. Total Synthesis of (±)-Moluccanic Acid Methyl Ester. Synlett. 2012;23(9):1358-1360.
29.
PempA.SeifertK. Enantioselective total synthesis of (+)-labd-8(17)-ene-3β,15-diol and (−)-labd-8(17)-ene-3β,7α,15-triol. Tetrahedron Lett. 1997;38(12):2081-2084. doi:10.1016/S0040-4039(97)00346-8
30.
KriegerJ.SmeilusT.SchackowO.GiannisA. Lewis acid mediated Nazarov cyclization as a convergent and enantioselective entry to C-nor-D-homo-Steroids. Chemistry. 2017;23(21):5000-5004.doi:10.1002/chem.201701008http://www.ncbi.nlm.nih.gov/pubmed/28345780
31.
SmithAB.LeenayTL. Indole diterpene synthetic studies. 5. Development of a unified synthetic strategy; a stereocontrolled, second-generation synthesis of (-)-paspaline. J Am Chem Soc. 1989;111(15):5761-5768.doi:10.1021/ja00197a039
32.
ViswanathanK.OnonyeSN.CooperHD.Kyle HaddenM.AndersonAC.WrightDL. Viridin analogs derived from steroidal building blocks. Bioorg Med Chem Lett. 2012;22(22):6919-6922.doi:10.1016/j.bmcl.2012.09.015http://www.ncbi.nlm.nih.gov/pubmed/23040731
33.
MajeedAJ.AntonsenØyvind.BennecheT.UndheimK. Stannylation reaction and cross-couplings in pyrimidines. Tetrahedron. 1989;45(4):993-1006.doi:10.1016/0040-4020(89)80011-0
34.
WangZ.XingZ.LiuLet al. Concise Total Synthesis of Isospongian Diterpenoid (±)-Polyrhaphin D. ChemistrySelect. 2016;1(10):2225-2227.doi:10.1002/slct.201600635
35.
YuJ.YuB. Synthesis of the ABC skeleton of the aglycon of echinoside a. Chin Chem Lett. 2015;26(11):1331-1335.doi:10.1016/j.cclet.2015.08.010
36.
GabrielaG.CarmenN.N-Tosyl-SFV. Sa)-binam-L-prolinamide as highly efficient bifunctional organocatalyst for the general enantioselective sol-vent-free Aldol reaction. Synlett. 2008;19:3031-3035.
37.
KirkDN.PetrowV.hormonesMsteroid.PartX. A new route to 4-methyl-3-oxo-D4-steroids. J Chem Soc. 1962:1091-1096.
38.
SakataK.WangY.UrabeD.InoueM. Synthesis of the tetracyclic structure of batrachotoxin enabled by bridgehead radical coupling and Pd/Ni-Promoted Ullmann reaction. Org Lett. 2018;20(1):130-133.doi:10.1021/acs.orglett.7b03482http://www.ncbi.nlm.nih.gov/pubmed/29232148
39.
SultanA.RazaAR.KhanKM. Towards the synthesis of batrachotoxin-formation of alkynyl stannanes. J Chem Soc Pakistan. 2013;33(5):1371-1385.
40.
AckermanLKG.LovellMM.WeixDJ. Multimetallic catalysed cross-coupling of aryl bromides with aryl triflates. Nature. 2015;524(7566):454-457.doi:10.1038/nature14676http://www.ncbi.nlm.nih.gov/pubmed/26280337
41.
LetortA.LongD-L.PrunetJ. Study of cascade ring-closing metathesis reactions en route to an advanced intermediate of taxol. J Org Chem. 2016;81(24):12318-12331.doi:10.1021/acs.joc.6b02264http://www.ncbi.nlm.nih.gov/pubmed/27978745
42.
MaC.LetortA.AouzalRet al. Cascade metathesis reactions for the synthesis of taxane and Isotaxane derivatives. Chemistry. 2016;22(20):6891-6898.doi:10.1002/chem.201600592http://www.ncbi.nlm.nih.gov/pubmed/27062670
43.
SmithAB.MewshawR. An efficient approach to chiral, nonracemic trans-decahydro-5,8a-dimethyl-1,6-naphthalenedione derivatives. Total synthesis of (+)-pallescensin A. J Org Chem. 1984;49(20):3685-3689.doi:10.1021/jo00194a003
44.
HagiwaraH.UdaH. Total synthesis of (+)-dysideapalaunic acid. J Chem Soc Chem Commun. 1988(12):815-817.doi:10.1039/c39880000815
45.
SwapnilN.KumarAS.BabuNJ.YadavJS. Synthetic approach towards the synthesis of antimalarial Gomphostenin. Asian J Org Chem. 2017;6(8):1091-1098.doi:10.1002/ajoc.201700131
46.
HaqueMA.SailoBL.PadmavathiG.KunnumakkaraAB.JanaCK. Nature-inspired development of unnatural meroterpenoids as the non-toxic anti-colon cancer agents. Eur J Med Chem. 2018;160:256-265.doi:10.1016/j.ejmech.2018.08.088http://www.ncbi.nlm.nih.gov/pubmed/30368201
47.
SnitmanDL.TsaiM-Y.WattDS.EdwardsCL.StotterPL. Convenient syntheses of 5,5,9-trimethyl-trans-1-decalone and 6.beta.-hydroxy-5,5,9.beta.-trimethyl-trans-1-decalone. J Org Chem. 1979;44(16):2838-2842.doi:10.1021/jo01330a004
48.
MukaiyamaT.MatsuoJ-ichi.KitagawaH.MatsuoJ.NewA. A New and One-Pot Synthesis of α,β-Unsaturated Ketones by Dehydrogenation of Various Ketones with N-tert -Butyl Phenylsulfinimidoyl Chloride. Chem Lett. 2000;29(11):1250-1251.doi:10.1246/cl.2000.1250
49.
DetheDH.MahapatraS.SauSK. Enantioselective Total Synthesis and Assignment of the Absolute Configuration of the Meroterpenoid (+)-Taondiol. Org Lett. 2018;20(9):2766-2769.doi:10.1021/acs.orglett.8b00997http://www.ncbi.nlm.nih.gov/pubmed/29672071
50.
IwasakiK.WanKK.OppedisanoA.CrossleySWM.ShenviRA.SimpleSRA. Simple, chemoselective hydrogenation with thermodynamic stereocontrol. J Am Chem Soc. 2014;136(4):1300-1303.doi:10.1021/ja412342ghttp://www.ncbi.nlm.nih.gov/pubmed/24428640
51.
TaguchiH.TanakaS.YamamotoH.NozakiH. A new synthesis of α,β-unsaturated aldehydes including (e)2-methyl-2-alkenal. Tetrahedron Lett. 1973;14(27):2465-2468.doi:10.1016/S0040-4039(01)96179-9
52.
MurokawaT.EnomotoM.TeranishiT.OguraY.KuwaharaS. Total synthesis of JBIR-03 and asporyzin C. Tetrahedron Lett. 2018;59(46):4107-4109.doi:10.1016/j.tetlet.2018.10.009
53.
MewshawRE.TaylorMD.SmithAB. Indole diterpene synthetic studies. 2. First-generation total synthesis of (-)-paspaline. J Org Chem. 1989;54(14):3449-3462.doi:10.1021/jo00275a035
54.
NakazakiK.HayashiK.HosoeS.TashiroT.KuseM.TakikawaH. First synthesis of decaturin C, an antiinsectant diterpenoid isolated from Penicillium thiersii. Tetrahedron. 2012;68(44):9029-9034.doi:10.1016/j.tet.2012.08.067
55.
KarimovRR.TanDS.GinDY. Synthesis of the hexacyclic triterpene core of the jujuboside saponins via tandem Wolff rearrangement-intramolecular ketene hetero-Diels-Alder reaction. Tetrahedron. 2018;74(26):3370-3383.doi:10.1016/j.tet.2018.04.051http://www.ncbi.nlm.nih.gov/pubmed/30467444
56.
Le ChapelainC. Strategy towards the enantioselective synthesis of schiglautone a. Org Biomol Chem. 2017;15(29):6242-6256.doi:10.1039/C7OB00766Chttp://www.ncbi.nlm.nih.gov/pubmed/28702653
57.
FujiiA.HashiguchiS.UematsuN.IkariyaT.NoyoriR.RutheniumNR. Ruthenium(II)-Catalyzed Asymmetric Transfer Hydrogenation of Ketones Using a Formic Acid−Triethylamine Mixture. J Am Chem Soc. 1996;118(10):2521-2522.doi:10.1021/ja954126l
58.
StymiestJL.BagutskiV.FrenchRM.AggarwalVK. Enantiodivergent conversion of chiral secondary alcohols into tertiary alcohols. Nature. 2008;456(7223):778-782.doi:10.1038/nature07592http://www.ncbi.nlm.nih.gov/pubmed/19079057
59.
WernerB.KalesseM. Pinacol Coupling Strategy for the Construction of the Bicyclo[6.4.1]tridecane Framework of Schiglautone A. Org Lett. 2017;19(7):1524-1526.doi:10.1021/acs.orglett.7b00288http://www.ncbi.nlm.nih.gov/pubmed/28300417
60.
XiaoW-L.LiR-T.HuangS-X.PuJ-X.SunH-D. Triterpenoids from the Schisandraceae family. Nat Prod Rep. 2008;25(5):871-891.doi:10.1039/b719905hhttp://www.ncbi.nlm.nih.gov/pubmed/18820756
61.
PengS-Z.ShaC-K. Stereoselective total syntheses of Guanacastepenes N and O. Org Lett. 2015;17(14):3486-3489.doi:10.1021/acs.orglett.5b01498http://www.ncbi.nlm.nih.gov/pubmed/26151193
62.
One of reviewers suggested following paper, which was published during preparation of present paper. Evanno L. Belotti D. Toromanoff E. Cossy J. synthesis of 12-epi-protopanaxadiol and formal synthesis of ginsenoside chikusetsusaponin-LT8. Eur J Org Chem. 2019;2019(34):5970-5973.