
Abstract
Epothilone D, a microtubule-stabilizing macrolide, is an attractive synthetic target molecule as a potential anti-cancer drug candidate. As part of our ongoing synthetic studies of this natural product, this paper describes the synthesis of the thiazole-containing northern segment of epothilone D via an E-selective bromomethylenation and a Ni/Cr-mediated cross-coupling reaction.
Epothilones are 16-membered macrolides isolated from the myxobacterium Sorangium cellulosum that have been identified as microtubule-stabilizing agents (Figure 1). 1 -3 Accordingly, they have attracted the attention of numerous researchers aiming to develop new anti-cancer drugs based on the structures of epothilones. 4 -7

Structures of epothilones A-D.
Their complex molecular architectures contain a thiazole ring and allow for a diverse range of synthetic strategies, and several successful total syntheses have been accomplished to date. 8
Among these natural products, epothilone D (
Intrigued by its molecular structure and promising biological activities, we decided to undertake a synthetic study of epothilone D. In this communication, we report the synthesis of a thiazole-containing northern segment of epothilone D.
Our retrosynthetic analysis of epothilone D is outlined in Scheme 1. Epothilone D was retrosynthetically divided into the northern and southern segments (

Retrosynthetic analysis of epothilone D (4).
Our synthesis commenced with the preparation of vinyl bromide
Horner-Wadsworth-Emmons Reaction Between 9 and 10.


The E:Z ratio was determined via 1H NMR analysis of the crude product.
The stereochemistries of the products were determined by downfield shift of the olefinic proton signal of (Z)
The resulting ester moiety of

Synthesis of allyl alcohol 13.
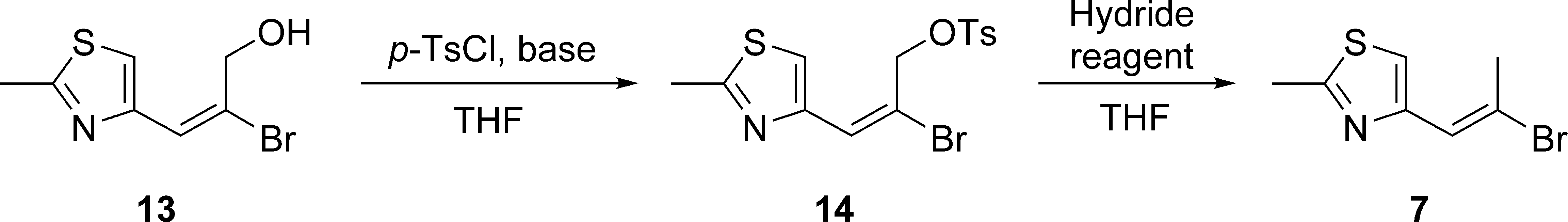
Conversion of Allyl Alcohol 13 to Vinyl Bromide 7.

Next, turning our attention to the coupling partner of

Synthesis of aldehyde 8.
With 2 segments
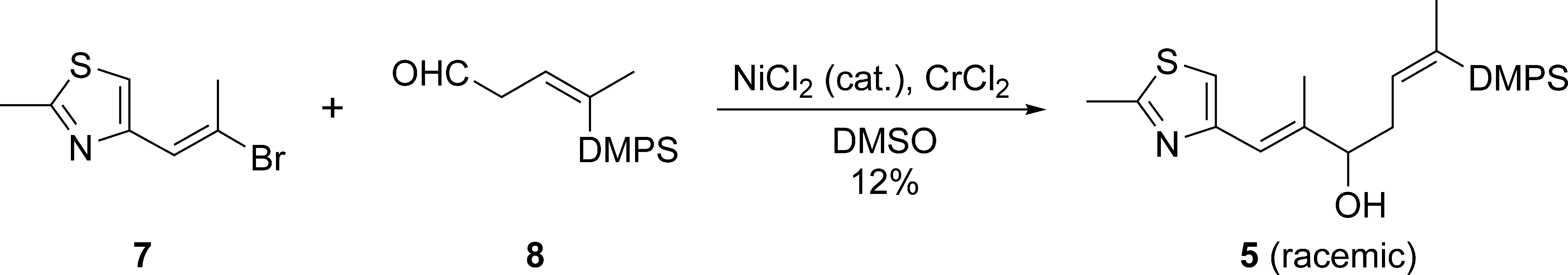
Ni/Cr-mediated coupling of 7 and 8.
In conclusion, we accomplished the synthesis of the thiazole-containing northern segment (
Studies toward the asymmetric synthesis of
Experimental
1H nuclear magnetic resonance (NMR) spectra were recorded on JEOL JNM-AL300 (300 MHz) instrument. The chemical shifts are expressed in ppm relative to tetramethylsilane (δ=0) as an internal standard (CDCl3 solution). Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; q, quartet; quint, quintet; m, multiplet; and br, broad peak. 13C NMR spectra were recorded on JEOL JNM-AL400 (100 MHz) instrument. The chemical shifts are reported in ppm relative to the central line of the triplet at 77.0 ppm for CDCl3. Infrared (IR) spectra were measured on a JASCO VALOR-III spectrometer and are reported in wavenumbers (cm−1). Both low-resolution mass spectra (MS) and high-resolution mass spectra (HRMS) were obtained using JEOL JMS 700 (double-focusing mass spectrometer [DFMS], electron impact ionization [EI], or fast atom bombardment [FAB] mode) instrument with a direct inlet system. Column chromatography was performed on silica gel (40-100 mesh). Analytical thin-layer chromatography (TLC) was conducted using 0.25 mm silica gel 60 F plates.
(E)-Methyl 2-Bromo-3-(2-Methylthiazol-4-yl)Acrylate (12)
To a stirred solution of
Rf: 0.33 (toluene/CH2Cl2, 5:1).
IR (CHCl3): 2953, 1732, 1434, 1359, 1224, 787 cm−1.
1H NMR (300 MHz, CDCl3): 2.75 (3H, s), 3.90 (3H, s), 8.34 (1H, s), 8.46 (1H, s).
13C NMR (100 MHz, CDCl3): 19.1, 52.9, 111.4, 119.7, 129.7, 149.0, 165.4, 165.5.
MS (EI, 70 eV): m/z = 263, 261 [M+].
HRMS-EI: m/z [M+] calcd for C8H8BrNO2S: 260.9459; found: 260.9456.
(E)-2-Bromo-3-(2-Methylthiazol-4-yl)-2-Propen-1-Ol (13)
To a stirred solution of ester
Rf: 0.43 (hexane/EtOAc, 5:1).
IR (CHCl3): 3232, 3019, 1508, 1189, 728 cm−1.
1H NMR (300 MHz, CDCl3): 2.73 (3H, s), 4.65 (2H, d, J = 7.3 Hz), 5.96 (1H, t, J = 7.3 Hz), 6.94 (1H, s), 7.02 (1H, s).
13C NMR (100 MHz, CDCl3): 19.1, 67.0, 118.0, 125.8, 128.6, 150.4, 167.1.
MS (EI, 70 eV): m/z = 235, 233 [M+].
HRMS-EI: m/z [M+] calcd for C7H8BrNOS: 232.9510; found: 232.9508.
(E)-2-Bromo-3-(2-Methylthiazole-4-yl)-2-Propene (7)
To a stirred solution of alcohol
To a stirred solution of the crude tosylate
Rf: 0.44 (hexane/EtOAc, 5:1).
IR (CHCl3): 2927, 1640, 1507, 1377, 1185, 788 cm−1.
1H NMR (300 MHz, CDCl3): 2.70 (3H, s), 2.77 (3H, d, J = 1.3 Hz), 6.85 (1H, m), 6.91 (1H, s).
13C NMR (100 MHz, CDCl3): 19.3, 30.3, 116.6, 125.1, 126.1, 151.5, 165.3.
MS (EI, 70 eV): m/z = 219, 217 [M+].
HRMS-EI: m/z [M+] calcd for C7H8BrNS: 216.9561; found: 216.9558.
4-(2-Tetrahydropyranoxy)-1-Butyne (15)
To a stirred solution of 3-butyn-1-ol (7.76 g, 111 mmol) in CH2Cl2 (100 mL) were added p-TsOH·H2O (212 mg, 1.11 mmol) and 3,4-dihydro-2H-pyran (10.3 g, 122 mmol) at 0°C. After stirring at the same temperature for 15 minutes, the reaction mixture was allowed to warm to room temperature and stirred for 3 hours. MeOH (10 mL) was added and the mixture was stirred for 30 minutes. The reaction was quenched by the addition of saturated aqueous NaHCO3 (50 mL) and the resulting mixture was extracted with CH2Cl2 (100 mL, 3 times). The combined organic layers were washed with brine (100 mL), dried over MgSO4, filtered, and concentrated in vacuo. The crude residue was purified via flash chromatography on silica gel (hexane/EtOAc, 5:1) to afford THP ether
Rf: 0.43 (hexane/EtOAc, 5:1).
1H NMR (300 MHz, CDCl3): 1.24-1.83 (6H, m), 1.98 (1H, t, J = 2.7 Hz), 2.50 (2H, dt, J = 7.1, 2.7 Hz), 3.50-3.62 (2H, m), 3.80-3.93 (2H, m), 4.65 (1H, t, J = 2.9 Hz). The spectroscopic data for
1-Dimethylphenylsilyl-4-(2-Tetrahydropyranoxy)-1-Butyne (16)
To a stirred solution of THP ether
Rf: 0.29 (hexane/EtOAc, 10:1).
1H NMR (300 MHz, CDCl3): 0.39 (6H, s), 1.51-1.72 (6H, m), 2.59 (2H, t, J = 7.1 Hz), 3.48-3.62 (2H, m), 3.82-3.90 (2H, m), 4.67 (1H, t, J = 3.3 Hz), 7.34-7.38 (3H, m), 7.61-7.65 (2H, m).
MS (FAB): m/z = 289 [M+H+].
(E)-1-Bromo-1-Dimethylphenylsilyl-4-(2-Tetrahydropyranyloxy)-1-Butene (17)
To a stirred solution of THP ether
Rf: 0.31 (hexane/EtOAc, 10:1).
IR (CHCl3): 2950, 1604, 1442, 1353, 1252, 1032 cm−1.
1H NMR (300 MHz, CDCl3): 0.55 (6H, s), 1.52-1.83 (6H, m), 2.19 (2H, dt, J = 7.9, 6.6 Hz), 3.26 (1H, dt, J = 9.5, 6.6 Hz), 3.43-3.49 (1H, m), 3.62 (1H, dt, J = 9.7, 6.4 Hz), 3.74-3.80 (1H, m), 4.60 (1H, t, J = 3.1 Hz), 6.80 (1H, t, J = 7.9 Hz), 7.34-7.40 (3H, m), 7.57-7.61 (2H, m).
MS (EI, 70 eV): m/z = 370, 368 [M+].
HRMS-EI: m/z [M+] calcd for C17H25BrO2Si: 368.0807; found: 368.0804.
(Z)-2-Dimethylphenylsilyl-5-(2-Tetrahydropyranyloxy)-2-Pentene (18)
To a stirred solution of vinyl bromide
Rf: 0.48 (hexane/EtOAc, 10:1).
IR (CHCl3): 2952, 1619, 1428, 1251, 1031, 816, 702 cm−1.
1H NMR (300 MHz, CDCl3): 0.40 (6H, s), 1.46-1.81 (6H, m), 2.29 (2H, q, J = 6.8 Hz), 3.26 (1H, dt, J = 9.5, 6.6 Hz), 3.43-3.49 (1H, m), 3.62 (1H, dt, J = 9.7, 6.4 Hz), 3.74-3.80 (1H, m), 4.60 (1H, t, J = 3.1 Hz), 6.80 (1H, t, J = 7.9 Hz), 7.34-7.40 (3H, m), 7.57-7.61 (2H, m).
13C NMR (100 MHz, CDCl3): −0.4 (2C), 20.3, 26.0, 26.3, 31.5, 33.5, 62.9, 67.9, 99.4, 128.6 (2C), 129.2, 129.6 (2C), 135.6, 140.2, 140.9.
MS (EI, 70 eV): m/z = 304 [M+].
HRMS-EI: m/z [M+] calcd for C18H28O2Si: 304.1859; found: 304.1862.
Anal. Calcd for C18H28O2Si: C, 71.00; H, 9.27. Found: C, 71.19; H, 9.35.
(Z)-4-Dimethylphenylsilyl-3-Penten-1-ol (19)
To a stirred solution of THP ether
Rf: 0.37 (hexane/EtOAc, 3:1).
IR (CHCl3): 3436, 2956, 1618, 1428, 1251 cm−1.
1H NMR (300 MHz, CDCl3): 0.46 (6H, s), 1.77 (1H, br s), 1.90 (3H, t, J = 1.5 Hz), 2.26 (2H, q, J = 6.3 Hz), 3.51 (2H, t, J = 6.3 Hz), 6.14 (1H, tq, J = 6.3, 1.5 Hz), 7.38-7.40 (3H, m), 7.57‒7.59 (2H, m).
13C NMR (100 MHz, CDCl3): −1.4 (2C), 25.2, 35.3, 62.2, 127.7 (2C), 128.8, 133.6 (2C), 136.0, 139.2, 139.5.
MS (EI, 70 eV): m/z = 220 [M+].
HRMS-EI: m/z [M+] calcd for C13H20OSi: 220.1283; found: 220.1280.
(Z)-4-Dimethylphenylsilyl-3-Pentenal (8)
To a stirred solution of alcohol
Rf: 0.44 (hexane/EtOAc, 5:1).
IR (CHCl3): 2960, 1720, 1428, 1223, 1111, 769 cm−1.
1H NMR (300 MHz, CDCl3): 0.40 (6H, s), 1.91 (3H, q, J = 1.3 Hz), 3.05 (2H, dquint, J = 7.5, 1.3 Hz), 6.22 (1H, tq, J = 7.5, 1.7 Hz), 7.31-7.39 (3H, m), 7.47-7.53 (2H, m), 9.45 (1H, t, J = 1.7 Hz).
13C NMR (100 MHz, CDCl3): −1.6 (2C), 25.3, 46.6, 128.1 (2C), 129.2, 131.8, 133.7 (2C), 138.5, 139.9, 199.9.
MS (EI, 70 eV): m/z = 218 [M+].
HRMS-EI: m/z [M+] calcd for C13H18OSi: 218.1127; found: 218.1129.
(1E,5Z)-6-Dimethylphenylsilyl-2-Methyl-1-(2-Methylthiazol-4-yl)-1,5-Heptadien-3-ol (5)
To a stirred solution of vinyl bromide
Rf: 0.1 (hexane/EtOAc, 5:1).
IR (CHCl3): 3341, 2929, 1617, 1508, 1428, 1210, 1110, 703 cm−1.
1H NMR (300 MHz, CDCl3): 0.41 (6H, s), 1.26 (1H, br s), 1.86 (6H, s), 2.29 (2H, t, J = 7.1 Hz), 2.70 (3H, s), 4.04 (1H, t, J = 6.2 Hz), 6.14 (1H, t, J = 6.2 Hz), 6.43 (1H, s), 6.90 (1H, s), 7.26-7.35 (3H, m), 7.51-7.55 (2H, m).
13C NMR (100 MHz, CDCl3): −1.3 (2C), 14.1, 19.2, 25.4, 37.9, 76.7, 115.5, 118.8, 127.9 (2C), 128.9, 133.7 (2C), 136.7, 139.3 (2C), 141.6, 152.8, 164.5.
MS (EI, 70 eV): m/z = 357 [M+].
HRMS-EI: m/z [M+] calcd for C20H27NOSSi: 357.1583; found: 357.1577.
Footnotes
Authors’ Note
This paper is dedicated to Professor Chiaki Kuroda on the occasion of his 65th birthday.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was partially supported by a grant from the Dementia Drug Resource Development Center, Project S1511016, the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.