
Abstract
This work deals with the isolation and pharmacological investigations of compounds of Euphorbia matabelensis. After multiple separation process, including thin layer chromatography (TLC), vacuum liquid chromatography, preparative TLC, and high-performance liquid chromatography, 1 diterpene (ingenol) and 2 flavonoids (naringenin and eriodictyol) were obtained from the methanol extracts prepared from the stems and roots of the plant. The structures of the isolated compounds were determined by nuclear magnetic resonance (NMR) and MS measurements and comparison with literature data. All compounds were isolated for the first time from the plant. Eriodictyol was detected for the first time from a Euphorbia species. The compounds were tested for their antiproliferative (on HeLa, C33a, MCF-7, and MDA-MB-231 cell lines) and GIRK channel blocking activities. None of the compounds proved to be active in these test systems.
The genus Euphorbia is one of the six largest genera (Astragalus, Bulbophyllum, Psychotria, Euphorbia, Carex, and Begonia) of flowering plants, with more than 2000 species worldwide, subdivided into many subgenera and sections. Members of this genus are characterized by the production of irritating milky latex. 1 For centuries, Euphorbia species have been used by various civilizations as sources of different medicines, due to their marked physiological effects. Diterpene-containing plants of the genus Euphorbia are of considerable interest as concerns natural product drug discovery programs because of the wide range of potentially valuable biological activities and broad structural diversity due to the different polycyclic and macrocyclic skeletons and various aliphatic and aromatic ester groups. 2 However, other compounds, eg, triterpenes, steroids, and flavonoids, also occur in these plants and can contribute to their diverse pharmacological activities. 3,4
Species belonging to the family Euphorbiaceae are known to contain skin irritant and cocarcinogenic compounds in the form of esters of the structurally related diterpenes, eg, tigliane, ingenane, and daphanane. 5 Many species of this family are used in folk medicine as drugs or as raw materials for medicinal preparations, eg, in traditional Chinese medicine they are recommended for the treatment of edema, gonorrhea, migraine, and warts. 2,6
Euphorbia matabelensis Pax (Euphorbiaceae) is a slightly succulent, deciduous shrub or tree, usually growing up to 3-8 m. The plant is harvested from the wild for local medicinal use and sometimes to make a gum. 7 In Malawi, a root decoction, combined with the leaves of Dichrostachys cinerea, is drunk to treat depression, high blood pressure, and swollen lymph glands. 7 In Zimbabwe, the powdered root is rubbed into scarifications on the breasts as a galactagogue for foster mothers. Moreover, the decoction of the chopped roots or latices is taken as a purgative in case of poisoning and to induce abortion, in which case a few drops are infused in a glass of milk and the infusion taken by orally. 7 The latex is taken into drinking water of chickens to treat diarrhea and Newcastle disease. 7
Until now, there is only one previous investigation of E. matabelensis, which was carried out by analyzing the latex of the plant. A diterpene of the ingenane-type parent alcohol with tetradecanoic acid ester as the acyl substituent was isolated by chromatographic methods from the latex of the plant. The ingenol ester exhibited irritant activity in mouse ear test. 8
As a part of our research program to discover new bioactive compounds from Euphorbia species, the chloroform-soluble fraction of a methanol extract of E. matabelensis Pax (Euphorbiaceae) was investigated. This study resulted in the isolation and structure determination of 1 diterpene (ingenol) and 2 flavonoids (naringenin and eriodictyol) from this plant. All compounds were studied for their antiproliferative activity against human tumor cell lines of gynecological origin, and GIRK channel-inhibitory activity using automated patch-clamp method, as previously several diterpene esters, isolated by our group, possessed such pharmacological activities. 9,10
The fresh plant material (root [R] and stem [S]) of E. matabelensis (1.6 kg [R] and 0.9 kg [S]) was chopped and extracted with methanol at room temperature. After concentration, the extracts were dissolved in 50% aqueous methanol, and solvent-solvent partitions were performed with CHCl3 and EtOAc. The CHCl3 phases were separated and fractionated with the combination of different chromatographic methods (column chromatography [CC], vacuum liquid chromatography [VLC], preparative thin layer chromatography [TLC], and high-performance liquid chromatography [HPLC]) to afford 3 compounds (Figure 1).
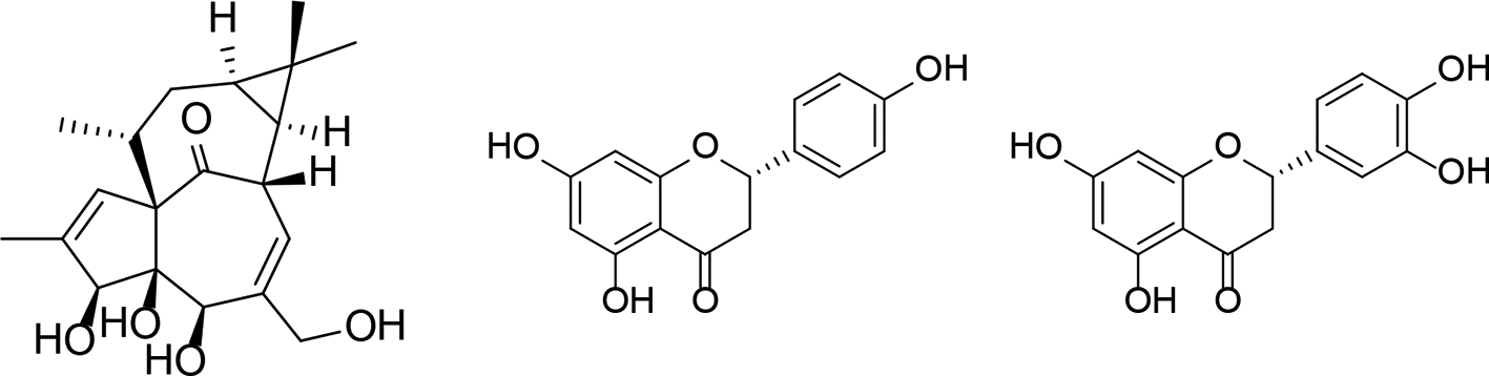
Structures of the isolated compounds 1 to
The structure determination was carried out by spectroscopic analysis, using 1- and 2-dimensional nuclear magnetic resonance (NMR) (correlated spectroscopy [1H-1H COSY], heteronuclear single-quantum correlation spectroscopy [HSQC], heteronuclear multiple-bond correlation spectroscopy [HMBC], and nuclear Overhauser enhancement spectroscopy) spectroscopy, high-resolution mass spectrometry, and comparison of the spectral data with literature data. Based on the NMR and MS investigations, compound
Ingenol has been detected previously in several Euphorbia species (eg, E. candelabrum, E. coralloides, E. deightonii, E. desmondii, E. drupifera, E. erythreaea, E. grandiflora, E. kamerunica, E. kotschyana, E. lactea, E. lathyris, E. memoralis, E. millii, E. nerifolia, E. nivulia, E. pentagona, E. pseudograntii, E. robbiae, E. royleana, and E. sikkimensis). 5
Ingenol 3-angelate, the best-known ingenol derivative, is used in human therapy (Picato) for the treatment of actinic keratosis, a precancerous skin disease. Moreover, it was reported that treatment of ingenol derivatives such as ingenol triacetate in HIV-infected T cells inhibited HIV replication through downregulation of CD4 receptors 14,15 and CXCR4 receptors. 16
Naringenin was isolated previously from different Euphorbia species, eg, E. sikkimensis, E. cuneata, and E. tuckeyana. 17 -19 The antioxidant potential of naringenin was confirmed in several in vitro studies and animal models. 20 Furthermore, the compound was found to be effective against the atypical multidrug resistant subline of gastric carcinoma and exhibited suppressive effect on transforming growth factor-β (TGF-β) signaling pathway, TGF-β ligand-receptor interaction; therefore, it could be of importance in several types of cancer wherein TGF-β plays an important role. Naringenin exerted cytotoxic activity against several tumor (eg, colon, pancreas, leukemia, stomach, cervix, liver, and breast) cell lines. 21
Eriodictyol was isolated for the first time from yerba santa (Eriodictyon californicum), a plant native to North America. 22 Eriodictyol and its glucoside possessed anti-inflammatory, anti-allergenic, antimicrobial, anticancer, and antioxidant properties. 23,24 Another study has confirmed that a dimer of naringenin and eriodictyol reduced brain damage and neurological deficits. 25 Moreover, eriodictyol enhanced insulin secretion dose-dependently only at high glucose concentration both in mice islets and MIN6 cells, improved glucose tolerance, and enhanced plasma insulin in nondiabetic and diabetic rats. 26
In our study, all compounds (
In conclusion, 3 compounds among them 1 diterpene (ingenol [
Experimental
General
CC was performed on polyamide (MP Biomedicals Germany GmbH, Germany). Normal phase VLC was carried out on silica gel (Kieselgel 60 GF254, 15 µm, Merck, United States). TLC was carried out on Kieselgel 60 RP-18 F254 and Kieselgel 60 F254 (Merck). Spots on UV active silica gel were detected under UV light (245 and 336 nm) and made visible with vanillin sulfuric acid and heating at 105°C for 2 minutes. The HPLC system was comprised of a Waters 600 controller, Waters 600 pump, and Waters 2998 photodiode array detector on an NP column (LiChroCART, 250-4, 5 µm). In case of the gradient elution, the mobile phase consisted of solvent A (cyclohexane) and solvent B (EtOAc) with a flow rate of 1.5 mL/min. The initial mobile phase composition was maintained at 80% solvent A for 1 minute, changed linearly to 20% (1-10 minutes) and held 2 minutes (10-11 minutes), then followed by a return to the initial conditions within 1 minute (11-12 minutes) and kept 2 minutes (12-14 minutes) for the chromatograph column equilibrium. The injection volume was of 25 µL. The data were acquired and processed with the Empower software.
NMR spectra were recorded in CD3OD on a Bruker Avance DRX 500 NMR spectrometer at 500 MHz (1H) and 125 MHz (13C). The signals of the deuterated solvents were taken as a reference. Two-dimensional experiments were performed with standard JEOL or standard Bruker software. In the COSY, HSQC, and HMBC experiments, gradient-enhanced versions were applied. The MS characterization was performed using an API2000 triple quadrupole mass spectrometer. The used ion source was turbo ESI source.
All solvents used for CC were of at least analytical grade (VWR Ltd., Szeged, Hungary). Ultrapure water was prepared with a Milli-Q water purification system (Millipore, France).
Plant Material
The stem and root (2.5 kg, fresh weight) of E. matabelensis Pax were collected in Kenya (Matuu subcounty, Machakos county S 01°04.579′ E 037°35.065′), Africa, in June 2016, and identified by Patrick Chalo Mutiso taxonomist. A voucher specimen (No. UON 2016/501) has been deposited at the Herbarium of the School of Biological Sciences, University of Nairobi, Kenya.
Extraction and Isolation
The fresh stems (S) and roots (R) of E. matabelensis (1.6 kg [S] and 0.9 kg [R]) were chopped and separately percolated with methanol (35 L) at room temperature. The crude methanol extracts were concentrated under reduced pressure (51 g [S] and 28 g [R]), and after solvent-solvent partition with CHCl3 and EtOAc, the CHCl3 phases were separated by polyamide column chromatography with MeOH-H2O gradient system (2:3, 3:2, 4:1, and 1:0) as eluent, to get four-four fractions (S-1-S-4 and R-1–R-4) from stems and roots. The fractions were monitored by TLC with the use of CHCl3-MeOH 95:5 as a mobile phase. After UV detection, the plate was sprayed with concentrated sulfuric acid and heated at 105°C for 2 minutes. Based on the TLC determination, it could be observed that fractions of stems and roots were differed from each other; therefore, their further purification was performed separately.
Fraction R1 (1.2 g) was separated by VLC on silica gel with a gradient system of CHCl3-MeOH (98:2, 95:5, 9:1, 8:2, 7:3, and 1:1; 100 mL of each). Fraction R1/4 (47 mg) was purified by preparative TLC on reversed-phase silica gel with MeOH-H2O 4:1 to yield compound
Fraction S2 (1.59 g) was separated by VLC on silica gel with a gradient system of cyclohexane-EtOAc-EtOH (9:1:0, 8:2:0, 7:3:0, 6:4:0, 60:30:3, 60:30:5, 6:3:1, and 1:1:1; 25 mL of each). Fraction S2/5 (11.6 mg) was subjected to NP-HPLC purification with gradient system of cyclohexane-EtOAc (flow rate 1.5 mL/min) to yield compound
Antiproliferative Assay
The antiproliferative properties of the isolated compounds were determined on a panel of human malignant cell lines isolated from cervical (HeLa and C33a) and breast carcinomas (MCF-7 and MDA-MB-231) purchased from European Collection of Cell Cultures (ECCAC, Salisbury, United Kingdom) exception for C33a cells which were from LGC Standards GmbH (Wesel, Germany). Cells were cultivated in minimal essential medium supplemented with 10% fetal bovine serum, 1% nonessential amino acids, and an antibiotic-antimycotic mixture. All media and supplements were obtained from Lonza Group Ltd. (Basel, Switzerland). Near-confluent cancer cells were seeded onto a 96-well microplate (5000 cells/well) and, after an overnight standing medium containing the tested compounds at 10 and 30 µM, was added. After incubation for 72 hours under cell culture conditions, the living cells were assayed by the addition of 20 µL of 5 mg/mL 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution. MTT was converted by intact mitochondrial enzymes and precipitated as purple crystals during a 4-hour contact period. The medium was next removed and formazan was dissolved in 100 µL of dimethyl sulfoxide (DMSO) during a 60-minute period of shaking at 37°C. Finally, the reduced MTT was assayed at 545 nm using a microplate reader. 27 All in vitro experiments were carried out on 2 microplates with 5 parallel wells. Stock solutions of the tested compounds (10 mM) were prepared in DMSO. The highest DMSO content of the medium (0.3%) did not have any substantial effect on the cell proliferation. Cisplatin (Ebewe Pharma GmbH, Unterach, Austria) was used as a reference agent.
GIRK Channel Inhibitory Assay
GIRK ion current was measured using planar patch-clamp technology in the whole-cell configuration with a 4 channel medium throughput fully automated patch-clamp system (Patchliner, Nanion Technologies GmbH, Munich, Germany). 28 The pipetting protocols were controlled by PatchControlHT 1.09.30 software (Nanion Technologies GmbH., Munich, Germany). Data acquisition and online analysis were performed with an EPC-10 Quadro patch-clamp amplifier (HEKA Elektronik Dr. Schulze GmbH, Lambrecht/Pfalz, Germany) using PatchMaster 2.65 software (HEKA Elektronik Dr Schulze GmbH, Lambrecht/Pfalz, Germany). Currents were low-pass filtered at 2.9 kHz using the internal Bessel filter of the amplifier and digitized at 10 kHz.
Experiments were carried out on HEK-293 (human embryonic kidney) cells stably expressing the GIRK1/4 (Kir3.1/3.4) K+ channels. 10 Cell line originated from UCL Business Plc. (London, Great Britain). Cells were maintained in Dulbecco's modified Eagle's medium (Thermo Fisher Scientific Inc., Waltham, United States) medium supplemented with 10% fetal bovine serum (PAN-Biotech GmbH, Aidenbach, Germany) and 182 µg/mL zeocin (Thermo Fisher Scientific Inc., Waltham, United States).
Automated patch-clamp experiments were carried out at room temperature with a suspension of stable transfected cell line. Suspension of cells for measurements was derived from running cell culture. Cells were maintained in incubator at 37°C, in 5% CO2. Before experiments, cells were washed twice with phosphate buffered saline (Thermo Fisher Scientific Inc., Waltham, United States) and then detached with trypsin-ethylenediaminetetraacetic acid (PAN-Biotech GmbH, Aidenbach, Germany) for 1 to 3 minutes. Trypsin was blocked with serum-containing media. The cell suspension was next centrifuged (2 minutes, 100 g), resuspended in serum-free media at a final density of 1 × 106-5 × 106 cells/mL, and kept in the cell hotel of the Patchliner. Cells were recovered after 15-30 minutes and remained suitable for automated patch-clamp recordings for up to 4 hours.
Stock of extra- and intracellular solutions were made for automated patch-clamp recordings. Chemicals were purchased from Sigma-Aldrich Corporation (St Louis, United States). All solutions were sterile filtered. Aliquots were stored at –20°C and warmed up to room temperature before use.
For each compounds isolated from E. matabelensis, a stock solution of test compound (10 mM) was prepared in each case. The solubilizing agent was DMSO (Sigma-Aldrich Corporation). Aliquots were stored at –20°C. Before experiments, stock solutions were further diluted with high K+ external solution to give appropriate concentrations for the measurements. The final DMSO concentrations in the tested samples were 0.1% or less.
At the beginning of the recordings, the normal external solution (4 mM K+) was replaced to high K+ external solution (25 mM K+) in order to increase the current amplitude. After 2-3 minutes of control period, the test compounds were added to the cells in increasing concentrations (1 and 10 µM), each for approximately 3 minutes. Propafenone (1 µM, Sigma-Aldrich Corporation) was used as a reference compound. Finally, potassium-free external solution was applied to completely cease inward potassium currents. The data were corrected with the current values measured in the potassium free external solution, which served as the baseline.
Footnotes
Acknowledgment
Financial support from the Economic Development and Innovation Operative Programme GINOP-2.3.2-15-2016-00012 is gratefully acknowledged. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is acknowledged.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was financially supported by the Economic Development and Innovation Operative Programme GINOP-2.3.2-15-2016-00012 and Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT.