
Abstract
Dental caries characterized by acid damage of tooth enamel is a persistent disease that begins with the formation of biofilms on the tooth surface. The secreted glucosyltransferases enable Streptococcus mutans to synthesize extracellular glucan polymers using ingested starch within the oral cavity, which eventually results in the production of acid, a contributing factor to cariogenesis. In this paper, we report the cloning, expression, purification, crystallization, and preliminary X-ray diffraction characterization of glucosyltransferase B.
Streptococcus mutans is a predominant pathogen that has been observed within the diseased sites in human dental caries. This pathogen helps shape the early biofilm and is responsible for the low pH (acidity), which is known to occur through its ability to ingest carbohydrates consumed by the host and convert them to glucan polymers. 1 Our group studies the structural and functional aspects of both the surface and extracellular proteins of S. mutans in order to understand the mechanism by which it promotes bacterial pathogenesis. Ultimately our goal is to interrupt the pathogenic role of S. mutans in dental caries through therapeutic interventions to prevent onset of the disease.
An important part of the formation of the biofilm is aided by the ability of S. mutans to produce glucan polymers from ingested sucrose and starches. These extracellular glucan polymers are manufactured by secreted enzymes collectively called the glucosyltransferases (Gtfs) as well as glucansucrases. 2 These glucan polymers are hypothesized to provide an additional platform for microbes to adhere and colonize, thus contributing to the construction of the biofilm community. The enzymatic activity of these exoenzymes creates a localized demineralizing acidic environment beginning the process of cariogenesis. 2 Among these Gtfs, GtfD produces only soluble alpha 1,6-glucan-linked polymers, GtfC produces both insoluble and soluble glucan polymers, whereas GtfB only produces insoluble glucan polymers. 3
While the crystal structure of GtfC has been resolved, very limited information has come forth in terms of the enzymatic mode of action, through which the Gtfs specifically produce the insoluble and soluble polymers. While GtfC has been shown to produce both the insoluble and soluble polymers, whether there is an allosteric site involved in suitably making the enzyme switch and/or if there are two distinct sites is still unknown. Sequence alignments of GtfB and GtfC show that they have 94% identity and 96% homology. Particularly in the GtfC’s active site, the residues are not very different from that of GtfB and GtfD. Given the peculiarities within these enzymes, and that to-date no specific enzymatic reaction mechanisms have been proposed and confirmed, we chose to structurally and functionally characterize GtfB. In this study we report the cloning, expression, purification, and crystallization of GtfB which encompasses the putative catalytic domain.
A construct of GtfB that includes the putative catalytic domain spanning residues 191 to 1051 was successfully cloned into pET-23d vector (Figure 1) and confirmed through DNA sequencing at the University of Alabama at Birmingham (UAB)-Heflin center for genomic science. The plasmids that harbor GtfB191-1051 were transformed into BL21(DE3) Escherichia coli cells, and after large-scale expression, the protein was first purified on an affinity HisTrap FF column where the sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis shows a predominate band around 100 kDa, as expected with a predicted MW of 96.3 kDa (Figure 2). Pooled protein was subsequently bound and purified over an anionic exchange MonoQ 10/100 column resulting in increased purity. Finally, the samples were run on a size exclusion Superdex 200 26/60 column rendering 99% purity as seen on the SDS-PAGE gel (Figure 2).

Primary structure of glucosyltransferase B (GtfB) showing the expressed construct with the catalytic domain, and the 6 33-residue glucan binding repeats (GBs) “WYYFDANGKAVTGAQTINGQTLYFDQDGKQVKG” that follow this domain.

Glucosyltransferase B (GtfB) was purified sequentially over three columns HisTrap FF, MonoQ HR10/100, and Superdex 200 26/60, and lanes respectively display the purified GtfB after each step.
Co-crystals of GtfB191-1051 with acarbose were obtained scanning for conditions using high-throughput crystallization with the ArtRobbins Gryphon robot. The Hampton Research Index Screen produced crystals in 2 M ammonium sulfate, 0.1 M Bis Tris, pH 5.5, shown both under visible and UV illumination in Figure 3. A complete dataset has been collected at SERCAT beamline 22-ID at the Advanced Photon Source in Chicago and the data collection parameters are listed in Table 1. A typical diffraction image for GtfB191-1051 is shown in Figure 4. The space group and unit cell dimensions are nearly identical to that of GtfC (Protein Data Bank [PDB]: 3AIC). Analogously, 8 molecules are expected in the asymmetric unit, where the Matthews’ coefficient of 4.3 corresponds to 73% solvent content rendering the crystals with poorer diffraction to 3.25 Å. 4 Using the GtfC structure as a model, we are currently using molecular replacement to resolve and refine this structure.
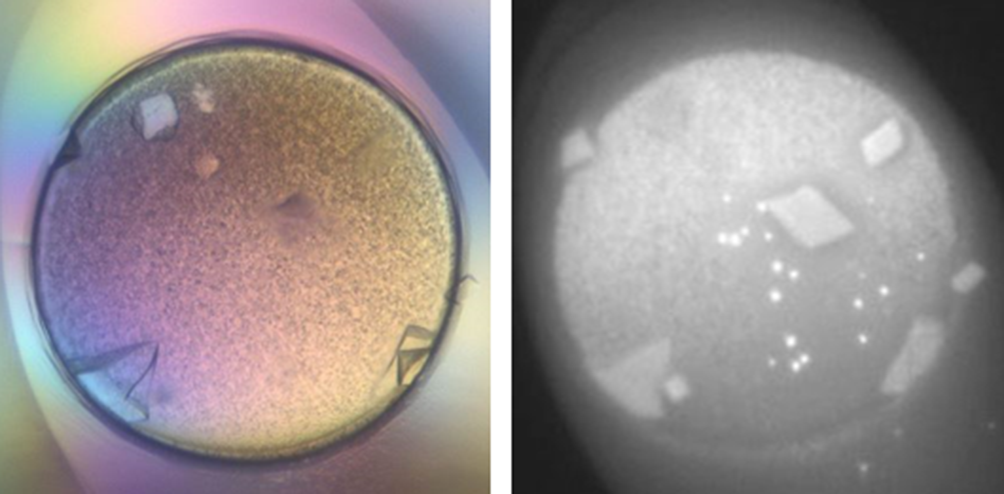
Glucosyltransferase B (GtfB191-1051) incubated with acarbose at a 1:10 molar ratio was screened using Index HT (Hampton Research). Crystals grew from condition A3 (2 M ammonium sulfate, 0.1 M Bis Tris, pH 5.5) after 1 week. The figure on the left is a visible image and on the right is the UV-illuminated image, which gave preliminary confirmation of these being protein crystals.
Crystallographic Data for GtfB191-1051.
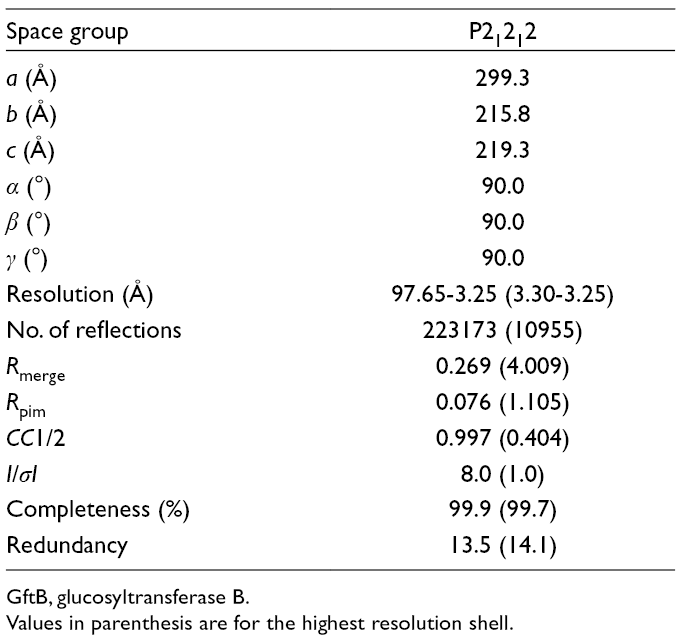
GftB, glucosyltransferase B.
Values in parenthesis are for the highest resolution shell.

Diffraction pattern of the glucosyltransferase B (GtfB191-1051) crystal collected at SERCAT on an Eiger detector.
Experimental
Molecular Cloning
Polymerase chain reaction (PCR) amplification of a fragment, GtfB191-1051, was carried out by Phusion HF DNA polymerase (New England Biolabs, Ipswich, MA, USA) using forward (“GGGG
Protein Expression and Purification
Escherichia coli BL21(DE3) cells harboring the GtfB191-1051 construct within the pET-23d vector were initially grown in 20 mL of Terrific broth (TB) in 100 mL shaker flasks overnight at 37°C and were transferred into 1 L of TB in 2400 mL shaker flasks the next day. Terrific broth cultures were then induced with 1 mM isopropyl β-d-1-thiogalactopyranoside at an optical density around 1.0 at 600 nm (OD600). Protein induction was continued for 5 hours at 30°C, and thereafter at 18°C overnight. These cells were pelleted by centrifugation (5000 × rcf for 20 minutes) using a Beckman Avanti J-25 centrifuge (Beckman Coulter, Brea, CA, USA). The pellets were then resuspended in the binding buffer (50 mM Tris pH 8.0, 500 mM sodium chloride, augmented with a complete ethylenediaminetetraacetic acid-free protease inhibitor cocktail) and sonicated in a Fisher Scientific Sonic Dismembrator 500 (Fisher Scientific, Hampton, NH, USA) for a total of 5 minutes while maintaining a maximum temperature of 10°C (using a temperature probe). The supernatant of the cell lysate was collected after centrifugation at 35,000 RPM for 1 hour on a Ti70 rotor, and subsequently filtered with a 0.22 µm filter and applied to a 5 mL HisTrap FF Column (GE Healthcare, Inc, Chicago, IL, USA). The bound GtfB was eluted with a gradient of 50 to 300 mM imidazole after thoroughly washing the unbound fractions with 50 mM imidazole in the binding buffer. The purest protein fractions were identified on SDS-PAGE gels, then pooled together and dialyzed overnight into 20 mM Tris pH 8.0 and 50 mM sodium chloride, and subsequently loaded onto a MonoQ 10/100 column (GE Healthcare, Inc). The MonoQ column fractions were dialyzed in the buffer containing 20 mM Tris pH 8.0 and 150 mM NaCl and finally loaded onto a Superdex 200 26/60 column (GE Healthcare, Inc). The purest fractions identified from SDS-PAGE gels were then concentrated under 55 psi nitrogen gas pressure using an Amicon stirred cell concentrator (MilliporeSigma, Burlington, MA).
Crystallization and Data Collection
The GtfB191-1051 (861 residues, MW 96.3 kDa) protein (at 15.8 mg/mL; 200 µM) was incubated for 30 minutes with acarbose at a 1:10 molar ratio (final concentration 2 mM). This protein-ligand solution was then subjected to high-throughput crystallization (hanging drop method; Corning 96-well plate) using our Gryphon (Art Robbins, Sunnyvale, CA, USA) robot and commercial screens. Index HT (Hampton Research, Aliso Viejo, CA, USA) condition A3 (2 M ammonium sulfate, 0.1 M Bis Tris, pH 5.5) yielded suitable protein crystals after 7 days (Figure 3). The condition was repeated in a manual setup (hanging drops and sitting drops), where we were able to routinely harvest crystals.
Crystals were cryo protected with 20% ethylene glycol and stored frozen for remote data collection at the SERCAT beamline 22-ID of the Advanced Photon Source in Chicago. Using an Eiger detector (Dectris, Baden, Switzerland) we collected a complete dataset (800 frames; 200° oscillation range; 0.25° oscillation per frame). Data were processed using XDS. 4 The crystal parameters are very similar to the deposited structure of GtfC (PDB: 3AIC; see Table 1). 5
Footnotes
Acknowledgments
Dr Deivanayagam and his group dedicate this work in honor of Dr Rama Krishna, who is both a professional colleague and personal friend of Dr Deivanayagam. His tireless contributions to the nuclear magnetic resonance (NMR) facility at UAB have resulted in a world class facility and in NMR faculty gravitating to UAB to conduct structural biology studies.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: HW and CD were partially supported by NIH/NIDCR R01DE017954. JLM is a T-90 Fellow and is supported by NIH/NIDCR T-90DE022736-06.