
Abstract
We describe Bayesian Model Optimized Reference Correction (BaMORC), a software package that performs 13C chemical shifts reference correction for either assigned or unassigned peak lists derived from protein NMR spectra. BaMORC provides an intuitive command line interface that allows non-nuclear magnetic resonance (NMR) experts to detect and correct 13C chemical shift referencing errors of unassigned peak lists at the very beginning of NMR data analysis, further lowering the bar of expertise required for effective protein NMR analysis. Furthermore, BaMORC provides an application programming interface for integration into sophisticated protein NMR data analysis pipelines, both before and after the protein resonance assignment step.
Chemical shifts derived from protein nuclear magnetic resonance (NMR) spectra have a wide variety of uses including protein structure determination, 1,2 characterizing ligand binding, 3-5 and drug discovery and design. 6,7 However, deriving accurate chemical shift values requires the referencing of NMR spectra to a certain standard, typically an internal standard. 8,9 Due to human errors and a variety of experimental factors, 10,11 errors occur quite frequently in 13C protein NMR data. An estimated 40% of the entries in the biological magnetic resonance bank (BMRB) have referencing issues. 12 The resulting referencing discrepancies are highly problematic since prior methods for reference correction required either assignment and/or structure, 13,14 which are the exact downstream aims that reference correction is trying to target. This leads to a co-dependency between reference correction and NMR structure determination, crippling the progress of many protein NMR analyses.
We therefore developed the Bayesian model optimized reference correction (BaMORC) method 15 that helps non-expert scientists to detect and correct 13C Cα and Cβ chemical shifts, at the beginning of the protein NMR analysis process, when chemical shifts are unassigned. Here, we describe the BaMORC method implemented in an easy-to-use software package written in the R programming language. BaMORC uses a Bayesian model to estimate an amino acid frequency from Cα and Cβ chemical shift statistics inferred from the re-referenced protein chemical shift Database (RefDB), 12 with or without resonance assignment information. As shown in Figure 1, by optimizing the minimal between the actual amino acid frequency calculated from known protein sequence and an estimation based on the observed chemical shifts, BaMORC returns the reference correction value and re-referenced chemical shifts data. Figure 2 illustrates the required input and expected output generated by the BaMORC R package.

Overview of the (unassigned) BaMORC algorithm.

Input utilized and output generated by the BaMORC R package.
The BaMORC R package provides a command-line interface (CLI) for general use and an application programming interface (API) for users that are familiar with R programming, especially for use within an integrated development environment like RStudio. 16 As illustrated in Figure 2, the BaMORC R package can use the protein sequence and chemical shifts in a variety of unassigned and assigned formats including the NMR-STAR format utilized by the BMRB. As illustrated in Figure 2, the general row-based text format may be delimited by a comma or white space, but with the protein sequence on the first line followed by unassigned peaks or assigned Cα and Cβ chemical shift pairs on following rows.
Each input file is referred to as a “task” within a larger “job”. The BaMORC R package automatically interfaces with the registration, grouping and referencing algorithms to set up tasks and derive the most optimized correction values for a given input, and returns the corrected chemical shifts in csv format. The package can also accept a BMRB ID such as BMR 4020 as input to retrieve corresponding files from the BMRB web server, automatically parsing the file, correcting the referencing, and returning the same set of output as mentioned before.
We have evaluated BaMORC against 568 13C protein NMR datasets from the RefDB with 90% or higher completeness with respect to Cα and Cβ chemical shift assignments. Outputted reference correction values should match closely to 0 ppm, since each dataset from RefDB has been reference-corrected using protein structure information. With chemical shift assignments, BaMORC provides reference correction values within ±0.50 ppm for all datasets and within ±0.22 ppm for 90% of the datasets, representing a 90% confidence interval (CI) of 0.40 ppm (Figure 3). 15 This level of performance is superior to the prior state-of-the-art linear analysis of chemical shifts (LACS) method. 14

Comparison of assigned BaMORC to the LACS method.
However, in the real-world situation, 13C reference correction is most valuable before protein resonance assignments are known. This situation is what the BaMORC package was really designed to address. The unassigned BaMORC method has two major components, grouping and referencing correction. With an input peak list, the grouping algorithm will return a list of Cα and Cβ grouped peaks (spin systems) as output, which will be the input for the referencing correction algorithm, as shown in Figure 2. The grouping algorithm is a variance-informed DBSCAN algorithm that employs derived dimension-specific match tolerance values to group peaks into spin systems. A peak list registration step is used to derive the necessary match tolerance values. 17 In addition to the grouped peaks, the referencing correction component uses the JPred4 18 server to generate sequence-based secondary structure predictions and then calculates the reference correction.
Again we used the same 568 13C protein NMR datasets from the RefDB to evaluate the reference correction component of unassigned BaMORC, but without chemical shift assignments. As shown in Figure 4, the reference correction component of unassigned BaMORC provides reference correction values within ±0.45 ppm for 90% of the datasets, representing a 90% CI of 0.69 ppm. 15 This suggests that the unassigned BaMORC algorithm can achieve the same level of performance when handling unassigned 13C protein NMR peak list data. This level of real-world performance is demonstrated with a set of peak lists derived from solution NMR HN(CO)CACB spectra for 10 different proteins. In this real-world evaluation, unassigned BaMORC provided reference correction values all within ±0.40 ppm. 15

Unassigned BaMORC reference correction accuracy.
Experimental
Software
The Python programming language, version 3.6, is used for the grouping algorithm. The R programming language, version 3.4, is used for the BaMORC core component. The library dependencies are listed below:
Python Library Dependencies: Python (≥3.6), gcc (≥5.1)
R Library Dependencies: R (≥3.4), data.table, tidyr, DEoptim, httr, docopt, stringr, jsonlite, readr, devtools, RBMRB, BMRBr
Experimental Data Sources
We used data from the RefDB to derive chemical shift statistics within the BaMORC package. For testing and evaluation, we used datasets from the RefDB and experimental peak lists from a variety of sources.
Installation
To use the BaMORC package, users must first install the R 3.4.x (or higher version) and Python 3.6.x (or higher version) interpreters on their machine. For Linux distributions, this is typically accomplished through the distribution’s package management system. For other operating systems, installation may require a more manual procedure. R language is a language and environment for statistical computing. 19 The installation guide is located in the website [https://cran.r-project.org/web/packages/BaMORC/index.html] of the comprehensive R Archive Network [https://cran.r-project.org/]. Python language 20 can be install from this website [https://www.python.org/].
Installing BaMORC From the Command Line (Linux and Mac Only)
To use BaMORC, the user first needs to install the package from the GitHub or CRAN.
$ wget -q https://cran.r-project.org/src/contrib/BaMORC_<version > .tar.gz
$ sudo R CMD INSTALL BaMORC_<version > .tar.gz
Install From Command Line via R Console
$ R # to start R console
>install.packages(“BaMORC”)
Install From R Console
>install.packages(“BaMORC”)
Installing Unassigned BaMORC Dependencies
The unassigned BaMORC analysis requires the ssc (spin system creator) package, which includes a variance-informed implementation of the DBSCAN algorithm used for protein NMR spin system clustering. A docker container including the ssc package is required. Therefore, the user needs to install both docker and SSC docker image.
Install Docker from https://www.docker.com/products/docker-desktop.
Install SSC docker container after docker is installed by running following code:
>docker pull moseleybioinformaticslab/ssc.
The BaMORC Application Programming Interface
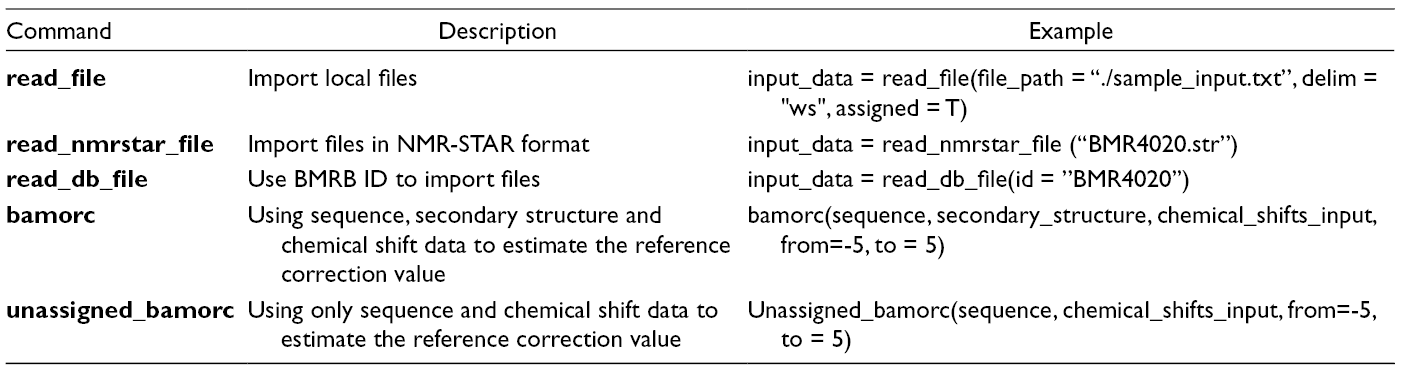
After importing the BaMORC in R either on R Console or in RStudio, the user will first read in NMR chemical shifts data via the read_file function with parameters of file path, file delimiter, and a flag that indicates whether data are either assigned or unassigned. BaMORC currently supports file delimiters of comma, semicolon, and whitespace. For users who want to run an analysis on an existing dataset from the BMRB (NMR-STAR version 2 and 3), they can use either the read_nmrstar_file function with a parameter for a local file path or the read_db_file function with a parameter for the BMRB ID and a flag that indicates whether data are assigned or unassigned. If read_db_file is used, BaMORC will utilize the BMRB web API to fetch the corresponding BMRB entry matching the ID. Table 1 shows common usage patterns for reading input data into the BaMORC referencing correction analysis pipeline. For a full list of available conversion options and more detailed examples and documentation of all the functions, please refer to “The BaMORC Reference” and “Quickstart.”
Summary of BaMORC Package Interface (API).
Next, the user will pass the input data as parameters to the bamorc() or unassigned_bamorc()function, which will perform the reference correction analysis. Both functions utilize the output from the read-in functions mentioned above and will perform a secondary structure estimation based on the provided protein sequence if secondary structure information is not provided. Through a series of optimization calculations (for details refer to paper 15 ), bamorc() and unassigned_bamorc() will return the estimated referencing correction value in a plain text file and corrected chemical shifts for both Cα and Cβ as a table, as shown in Figure 2. The user can optionally customize the search range. Table 1 contains a basic example of calling each function. For detailed examples and expected outputs of BaMORC API functions, refer to the online documentation: https://moseleybioinformaticslab.github.io/BaMORC/index.htm.
The BaMORC Command Line Interface
The BaMORC CLI is an extension of the BaMORC package, aimed at the broader NMR community that is not familiar with R programming language. To use BaMORC CLI, the user needs to find the CLI run-script first by opening a terminal and typing the command highlighted in Figure 5.
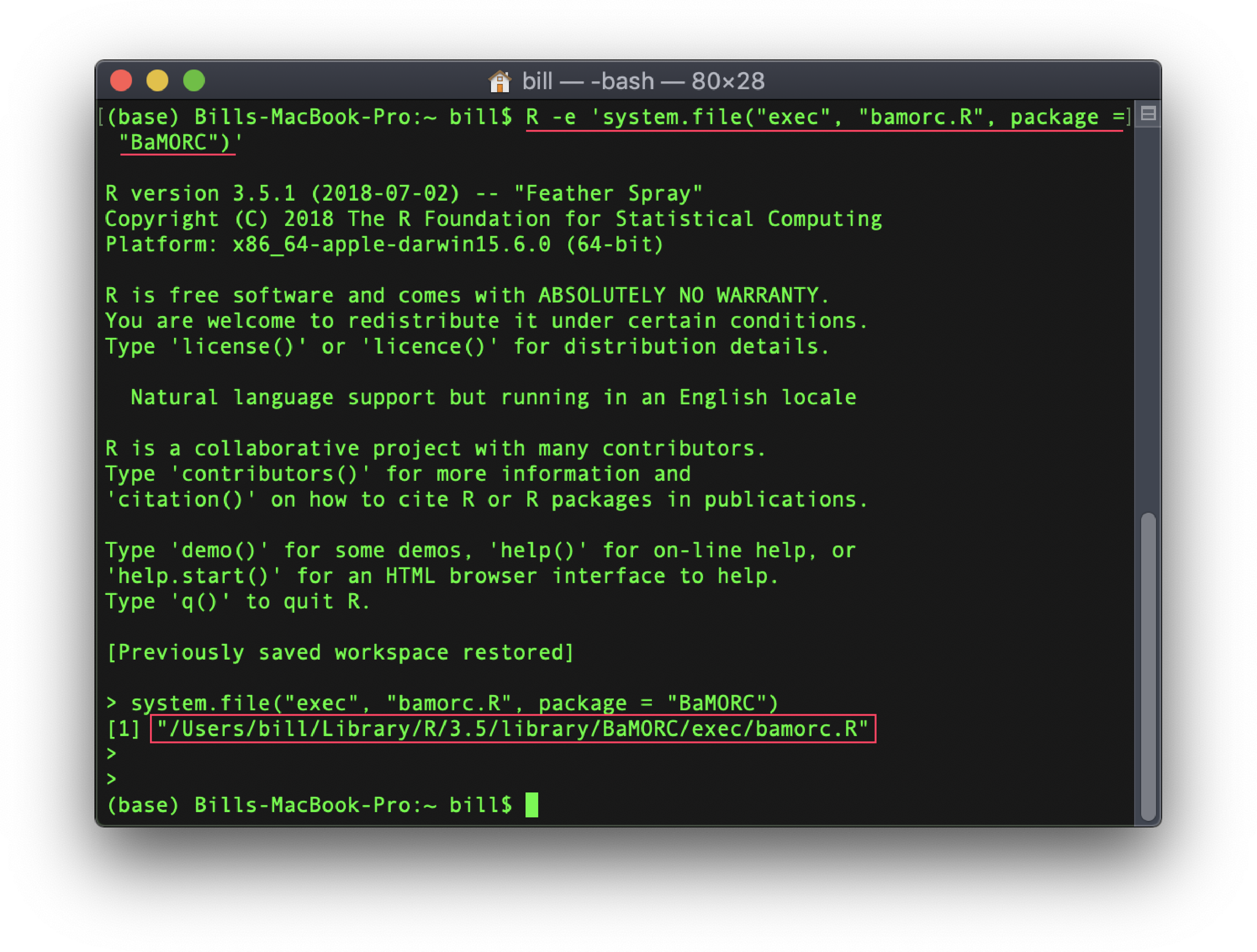
Finding the CLI run-script location.
>R e 'system.file("exec," "bamorc.R," package = "BaMORC")'
The user can then execute the appropriate command listed in Table 2 to run an analysis. Similar to the package, the BaMORC CLI has three major modules: assigned and unassigned reference correction for assigned and unassigned protein NMR data and a miscellaneous collection of other useful tasks. Table 2 lists the components of the CLI and their associated parameters.
BaMORC CLI Commands and Their Parameters.

To help the user transition between the API and CLI, Table 3 illustrates common BaMORC CLI usage examples with corresponding BaMORC API examples. The CLI is utilized within a command line terminal on Linux and Mac computers. For windows user, refer to our online documentation for more details.
BaMORC CLI Usage and Corresponding API Commands.

We have developed online documentations, available at: https://moseleybioinformaticslab.github.io/BaMORC/index.html.
Reporting Summary
Further information on the algorithms mentioned above and their development is available. 15
Code Availability
Source code is available at https://github.com/MoseleyBioinformaticsLab/BaMORC. [The package has been submitted to CRAN and should be available from CRAN soon. We will add a sentence about its availability from CRAN and update installation instructions when the evaluation process is finished]. The code is published under a modified open source BSD-3 license. Academic researchers are free to use it without restriction, except for proper citation. This repository includes code for the BaMORC referencing correction pipeline. For the registration and grouping algorithm, refer to https://github.com/MoseleyBioinformaticsLab/ssc. 21 For further information and assistance visit our laboratory website: http://bioinformatics.cesb.uky.edu.
Data Availability
Datasets are available at: https://doi.org/10.6084/m9.figshare.5270755.v1
Footnotes
Acknowledgments
The authors acknowledge support from the National Science Foundation grant and National Institutes of Health grants.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Support for this research was provided by the National Science Foundation grant NSF 1419282 (Hunter N.B. Moseley) and National Institutes of Health grants NIH UL1TR001998-01 (Philip Kern) and NIH P30CA177558 (Mark Evers).