
Abstract
Background:
A head-to-head trial (NCT03078478) between insulin degludec and insulin glargine U300 with the primary objective of comparing the risk of hypoglycemia is being conducted. During trial conduct, safety concerns related to the glycemic data collection system led to a postinitiation protocol amendment, described here.
Methods:
This randomized (1:1), open-label, treat-to-target, multinational trial was initiated in March 2017 with a planned treatment period of 52 weeks (16 weeks titration + 36 weeks maintenance). Overall, ~1600 insulin-experienced patients at risk of developing hypoglycemia based on predefined risk factors were included. The protocol amendment implemented in February 2018 resulted in assuring patient safety and an extension of the total treatment period up to 88 weeks (16 weeks titration + variable maintenance 1 + 36 weeks maintenance 2). The original glycemic data collection system (MyGlucoHealth blood glucose meter + electronic diary) was discontinued because of safety concerns and replaced with an Abbott blood glucose meter and paper diary to collect self-measured blood glucose and hypoglycemic episodes. The primary endpoint of number of severe or blood-glucose confirmed symptomatic hypoglycemic episodes will be evaluated with the same analysis duration and statistical methods as the original protocol. Only relevant changes were implemented to maintain patient safety while permitting evaluation of the scientific objectives of the trial.
Conclusions:
These observations highlight the importance of safety surveillance during trial conduct despite the use of currently marketed glucose monitoring devices. The prompt protocol amendment and ensuing actions ensured that the scientific integrity of the trial was not compromised.
Hypoglycemia remains a major barrier to initiating and intensifying insulin therapy for the management of type 2 diabetes (T2D).1,2 New longer-acting basal insulin analogues such as insulin degludec (degludec) and insulin glargine 300 units/mL (glargine U300) are recognized for their peakless pharmacodynamic profiles contributing to low variability in glucose-lowering effect compared to insulin glargine 100 units/mL (glargine U100).3-5 However, it is critical that the beneficial effects of these insulins outweigh the associated risk of hypoglycemia, especially severe and nocturnal episodes that are potentially life-threatening.2,6-8
Previously, it has been shown that glargine U300 has a more stable pharmacodynamic profile compared to glargine U100. 5 In comparison to both concentrations of glargine, degludec has lower day-to-day variability in glucose-lowering effects.3,9 Contrasting results were observed in two studies comparing degludec and glargine U300 for within-day variability possibly due to differences in the experimental design and statistical analyses.9,10,11 It can be expected that distinct pharmacodynamic profiles of these insulins differentially influence their clinical profiles including the risk of hypoglycemia. Several studies have shown that variability in glucose-lowering effect is associated with an increased risk of hypoglycemia and other diabetes-related complications.12-15 Furthermore, randomized controlled trials have demonstrated a difference in the risk of hypoglycemia between degludec, glargine U300 and glargine U100 while confirming noninferiority in HbA1c reduction.16-19
In order to examine the risk of hypoglycemia with degludec compared to glargine U300 in a clinical setting, a randomized active-controlled trial was initiated in March 2017 including ~1,600 T2D patients previously treated with basal insulin. In contrast to a recent study between glargine U300 and degludec having glycemic control as the primary endpoint, 19 this study was designed and powered to evaluate the superiority of degludec versus glargine U300 for the number of severe or blood glucose (BG)–confirmed (<56 mg/dL) symptomatic hypoglycemic episodes as the primary endpoint. During trial conduct, routine medical monitoring activities on blinded data revealed an unusual and potentially unsafe reporting pattern of glycemic values and hypoglycemic episodes. The data indicated that these patterns were related to the glycemic data collection system used in the trial which constituted the MyGlucoHealth (MGH) blood glucose meter (BGM) and an electronic diary (e-diary) to capture self-measured blood glucose (SMBG) values and patient-reported information related to hypoglycemia. Therefore, in the interest of patient safety, a major protocol amendment was implemented to discontinue and replace the glycemic data collection system. The primary aims of this article are to highlight the description and reasons for the protocol amendment while providing an overview of the data handling measures and statistical considerations to maintain the scientific integrity for this trial. In addition, the learnings from our investigation of the issue and the mitigation plan can provide helpful insights for similar issues arising in future trials.
Methods
Trial Design
This randomized, open-label, parallel, multicenter, multinational, treat-to-target, active-controlled trial is being conducted to show whether treatment with degludec (Tresiba®, Novo Nordisk, Bagsvaerd, Denmark) is associated with a lower rate of severe or BG-confirmed symptomatic hypoglycemia compared to glargine U300 (Toujeo®, Sanofi, Paris, France) when both treatments are administered once-daily, with or without oral antidiabetic drugs (OAD) in insulin-experienced T2D patients.
The original trial duration comprised 52 weeks of active treatment with 16 weeks of titration and 36 weeks of maintenance (Figure 1). The trial protocol was amended in February 2018 to include a new maintenance period 2 (M2) of 36 weeks resulting in total trial duration up to 94 weeks with active treatment up to 88 weeks (Figure 1). The rationale for this amendment is discussed in the following sections. The primary and secondary objectives of the trial remained unchanged following the protocol amendment (Figure 1).

Trial design pre- and postamendment.
Patients included in the trial were randomized 1:1 to degludec or glargine U300. Within each treatment arm, patients were also randomized 1:1 to morning (from waking-up to breakfast) or evening dosing (from main evening meal to bedtime) and the same dosing time will be retained throughout the treatment duration. The type and dose of pretrial OAD should be unchanged throughout the trial unless for safety reasons.
The trial is registered with clinicaltrials.gov (NCT03078478) and is being conducted in accordance with the Declaration of Helsinki and Good Clinical Practice Guidelines. Signed informed consent was obtained before initiating any trial-related activity. Prior to initiating activities related to M2, all patients were asked to reconsent in order to continue their participation in the trial.
Patients
The inclusion and exclusion criteria are listed in Table 1. The inclusion criteria are similar to the previously conducted SWITCH 2 trial (clinicaltrials.gov: NCT02030600) comparing degludec with glargine U100 in T2D patients. 16
Key Inclusion and Exclusion Criteria.

OAD includes any combination of metformin, dipeptidyl peptidase-4 inhibitor, α-glucosidase inhibitor, thiazolidinediones, and sodium glucose co-transporter-2 inhibitor at stable doses.
Glycemic Data Collection System
The Conformité Européenne (CE) and 510(k)-approved MGH BGM (Entra Health Systems LLC, California, US) was used to record SMBG values at trial commencement. SMBG values from the BGM were captured in an e-diary using wireless transfer technology, collectively called the glycemic data collection system. Patients received the manufacturer’s instructions for use and sites provided supplementary training at randomization and the following visit. The e-diary was also used by patients to report insulin doses and information related to hypoglycemic episodes, including the date, time, SMBG value, symptoms, and ability to self-treat.
During trial conduct, routine medical monitoring on raw blinded data showed an unusual pattern in the reporting of glycemic parameters and hypoglycemic episodes. To evaluate these findings the data were compared to those from SWITCH 2 16 that included a trial population similar to the current trial. In both these treat-to-target trials, insulin dose was up-titrated once-weekly based on the mean of three preceding daily prebreakfast SMBG values. In SWITCH 2, a standard Abbott BGM was used for SMBG measurements.
Glycemic data in this trial revealed an inconsistency between parameters measured at the central laboratory (HbA1c and fasting plasma glucose [FPG]) and the patient-reported SMBG values. In comparison to SWITCH 2, mean values of HbA1c and FPG were lower, while mean SMBG was higher (Figure 2). Thus, data available to the patient from SMBG monitoring were suspected to indicate that BG was higher than it was. As SMBG measurements were used for dose titration, this could have led to an increased risk of hypoglycemia, including severe episodes, since the patient would see values higher than the true value possibly resulting in an insulin dose higher than the actual dose required.
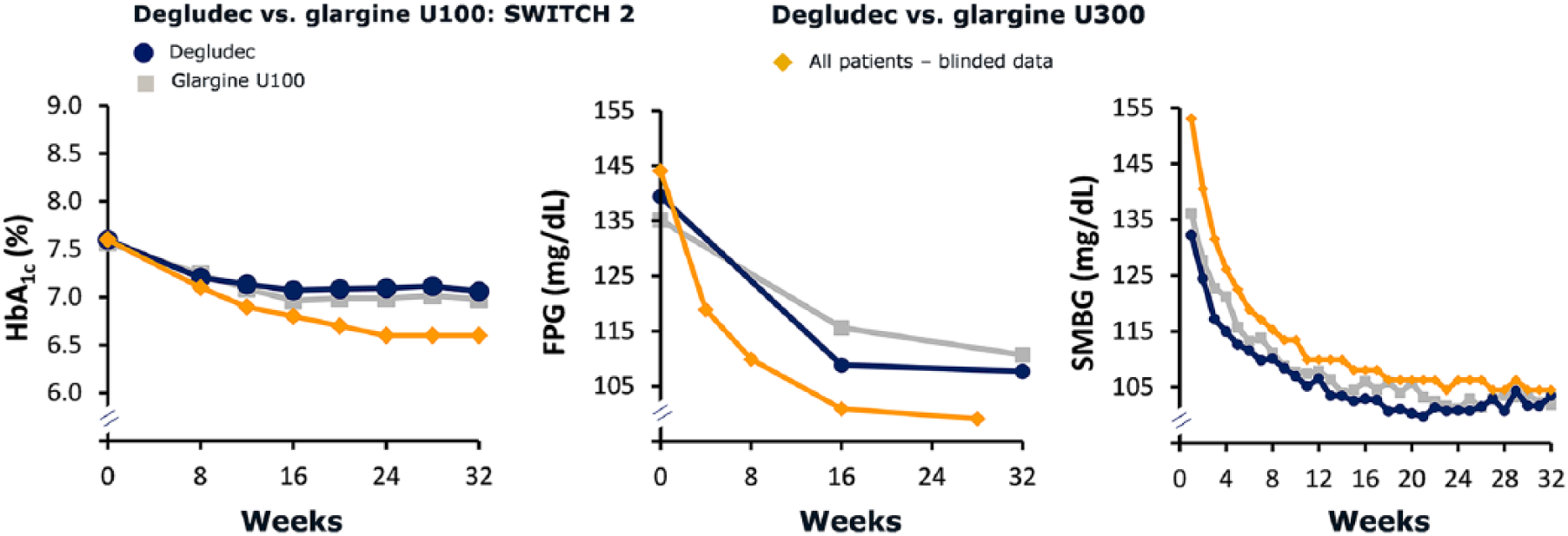
Glycemic parameters compared to SWITCH 2. Mean values presented.
In addition, when the protocol amendment was considered, the rate of patient-reported BG-confirmed hypoglycemic episodes (<56 mg/dL) was lower and the rate of symptomatic hypoglycemia with BG >70 mg/dL or without corresponding BG measurements was higher than those observed in SWITCH 2. 16 Importantly, a higher-than-expected proportion of patients had reported clinically serious symptoms related to severe hypoglycemia. Of note, all severe episodes during the total trial period will be evaluated by an external adjudication committee and at the time of the amendment no severe hypoglycemic episode had been adjudicated.
The evidence suggested that these data discrepancies observed as a trend across the entire trial population were related to the glycemic data collection system, thus mandating a major protocol amendment in the interest of patient safety.
Protocol Amendment Process
All procedures related to this protocol amendment were carried out with adherence to the Good Clinical Practice guidelines (ICH E6 R2). 21 Regulatory agencies, institutional review boards and ethics committees in all participating countries were notified about the safety concern and the protocol amendment, and approvals were obtained as relevant in accordance with local procedures. Since both trial products are marketed, there were no additional regulatory obligations due to increased drug exposure. Implementation of the protocol amendment commenced on February 14, 2018, by informing all investigators and trial sites to contact their patients and have them discontinue the MGH BGM and e-diary with immediate effect. At the time, recruitment had been finalized and all patients on-treatment had completed the titration phase using the MGH BGM and e-diary. The transition phase including the original maintenance period is considered maintenance period 1 (M1) (Figure 1), during which patients were asked to use their own BGM and a temporary paper diary. Patients continued their randomized treatment during the transition phase as they would have during the maintenance phase of the trial. The duration of M1 was dependent on each patient’s individual randomization date and/or receipt of approval from health authorities or local ethics committees, if applicable. Subsequently, patients were invited to reconsent and initiate M2 as soon as the required resources were available at the trial sites. At this visit all patients received a standard Abbott BGM and paper diary to be used through the remaining trial duration. The trial sites were supplied with detailed instructions on handling the amendment with minimal inconvenience to the patients, while ensuring ongoing ethical trial conduct.
Root-Cause Analysis
Following the amendment, the sponsor (Novo Nordisk) conducted an assessment of the MGH BGM and test strip lots by trial sites and countries to investigate whether a particular version of the meter or strip lot used by patients could have impacted the results. The assessments showed that there were no differences in the data collection pattern based on two BGM firmware versions (versions 3.11 and 3.02) used in the trial. The test strip lots were distributed evenly among trial sites. Due to dynamic improvements in BG levels, comparison of BG values between the strip lots used at different time points during the trials had limited value. Thus, it was not possible to detect any pattern of specific lots measuring BG values significantly different from the rest of the lots. The pairing of a specific test strip lot with a particular BGM is unknown since the test strip ID is not transferred to the database. No patterns in SMBG readings were observed based on the geographical locations of the trial sites. In addition, the accuracy of data transfer from the MGH BGM to the clinical trial database was confirmed.
The performance and accuracy of the MGH BGM and test strips were tested in an independent institute using laboratory tests and a clinical trial according to ISO 15197:2015 22 and FDA guidelines. 23 Results from these analyses have been described elsewhere.24,25
Endpoints
The primary and secondary confirmatory endpoints pre- and postamendment are listed in Table 2. The description and analysis duration for the primary endpoint of number of severe (episode requiring third-party assistance) 20 or BG-confirmed (<56 mg/dL) symptomatic hypoglycemic episodes during the maintenance period remains unchanged postamendment.
Endpoints Pre- and Postamendment of the Trial Protocol.
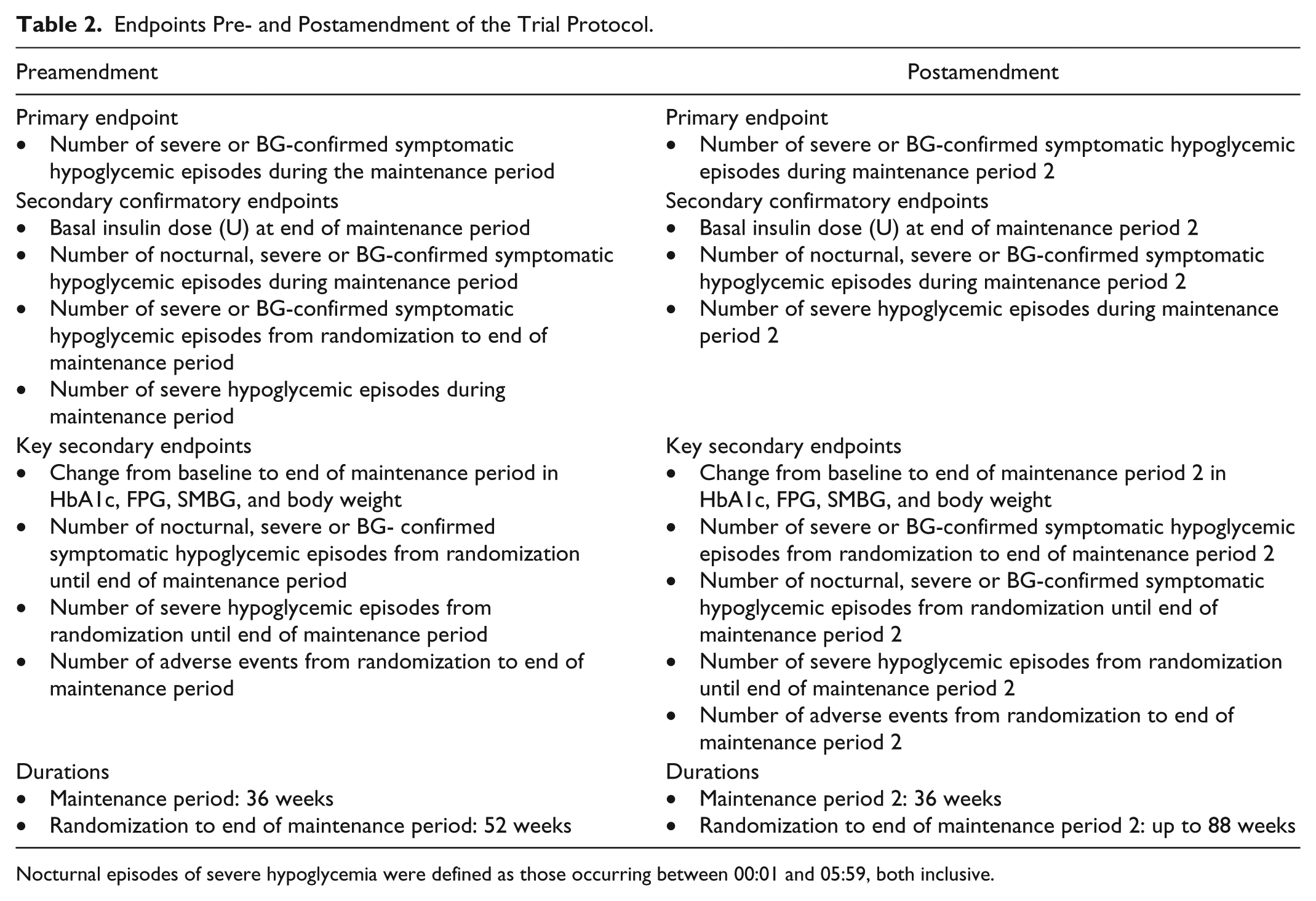
Nocturnal episodes of severe hypoglycemia were defined as those occurring between 00:01 and 05:59, both inclusive.
However, as a consequence of data collection from different systems during the total treatment period (Figure 1) the test hierarchy was modified to exclude the secondary confirmatory endpoint of number of severe or BG-confirmed symptomatic hypoglycemia during the total treatment period. This endpoint will be assessed as an additional secondary endpoint (Table 2).
Statistical Considerations
The primary and secondary confirmatory endpoints will be analyzed to investigate two aspects of the treatment effect. The primary analysis is the treatment difference between degludec and glargine U300 assuming that all patients adhered to the randomized treatment and is thus referred to as the on-treatment analysis. The on-treatment analysis was selected as the primary analysis with the aim of comparing the primary and secondary confirmatory endpoints related to hypoglycemia occurring due to exposure to the trial products.
The secondary analysis is the treatment difference between degludec and glargine U300 irrespective of treatment discontinuation or adherence to randomized treatment, thus following the intent-to-treat (ITT) principle. The ITT analysis addressing treatment effect in all randomized patients was selected to maintain the randomization integrity and eliminate any potential bias between treatment arms due to patients discontinuing the randomized treatment.
The sample size for this trial was calculated to ensure at least 80% power for the on-treatment analysis on the primary and secondary confirmatory endpoints (except for severe hypoglycemia, the last endpoint in the hierarchical testing procedure).
Endpoints related to hypoglycemia will be summarized using the safety analysis set (patients receiving at least one dose of the randomized treatment) for on-treatment data and full analysis set for in-trial data. All efficacy endpoints will be summarized using the full analysis set and other safety endpoints will be summarized using the safety analysis set.
The confirmatory endpoints will be tested in a hierarchical order using the on-treatment analysis to control the type 1 error in the strong sense. As shown in Table 3, confirmatory endpoints (except insulin dose) will be measured during M2 for the same analysis duration as that prior to the amendment. Statistical methodology related to the primary and secondary confirmatory endpoints are unchanged pre- and postamendment (Table 3). In order to evaluate the robustness of the primary analysis, sensitivity analyses will be performed as described in Table 4. Patients discontinuing the randomized treatment prior to M2 will be characterized to evaluate any potential imbalance between the treatment arms based on demographics and baseline characteristics.
Statistical Analyses for Confirmatory Endpoints.
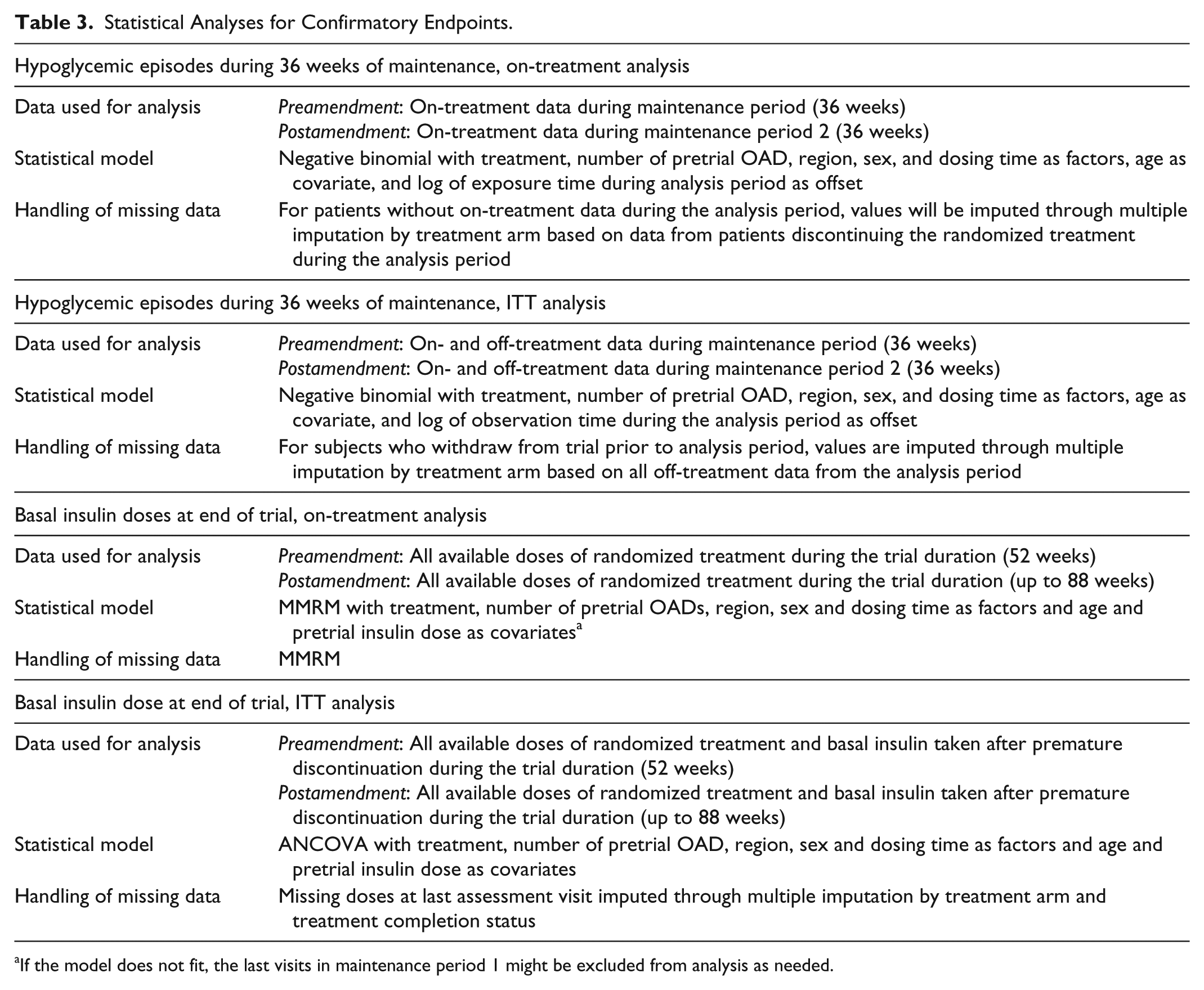
If the model does not fit, the last visits in maintenance period 1 might be excluded from analysis as needed.
Sensitivity Analyses for the On-treatment Analysis.

Following the protocol amendment, patients were asked to reconsent in order to continue their participation in the trial.
Last, to supplement the conclusions on the primary and secondary confirmatory endpoints, additional endpoints including HbA1c, FPG, SMBG, and adverse events will be assessed (Table 2). These endpoints evaluated during the entire trial duration will be reported in accordance with the original protocol plan.
Discussion
During trial conduct of a randomized controlled trial comparing degludec versus glargine U300, primarily with a focus on hypoglycemia in insulin-experienced patients with T2D, unusual reporting of glycemic data was observed. This imposed a significant possibility of exposing patients to a high risk of severe hypoglycemic episodes. Based on all information available, this anomalous data reporting pattern was attributed to the glycemic data collection system constituting the MGH BGM and e-diary. Reports of inaccurate BG values measured using standard approved BGMs are unfortunately not unique to this trial. Previously, several studies have concluded that marketed BGMs deviate from the minimum accuracy requirements specified by international standards.26-28 In some cases the deviations were attributed to variability between BGM test strip lots. 29 These reports and findings from the current trial highlight the importance of accuracy and reliability in BG monitoring to preclude false treatment decisions that could impose a threat to patient well-being. Of note, the MGH BGM was approved by regulatory bodies yet failed to report data accurately, which may indicate a need for ongoing intermittent surveillance of marketed BGM systems. It is of importance to note that the medical monitors and safety surveillance advisors evaluating these data were blinded to the treatment and the sponsor’s safety committee based their conclusions solely on the raw data reported.
The postinitiation protocol amendment of this trial was deemed obligatory to safeguard trial participants; however, it did not change the key objectives and features of the trial. The sponsor conducted a thorough assessment to ensure that only critical changes were implemented in order to minimize inconvenience to patients and trial site personnel while maintaining trial integrity.
The treat-to-target trial design was selected with the aim of achieving similar glycemic control in both treatment arms that would enable appropriate assessment of the risk of hypoglycemia. In addition, analysis of the primary endpoint during the maintenance period is considered appropriate due to relatively stable insulin doses and similar levels of glycemic control expected between the treatment arms during this period compared to the titration period. This approach is also clinically relevant to real-life scenarios in which stable doses of basal insulin are often administered for long-term diabetes treatment. The maintenance period was thus replicated as is following the protocol amendment. As shown in Figure 1, different glycemic data collection systems will be used during the trial that could possibly lead to discrepancies in the BG and hypoglycemia data collected. Hence, hypoglycemia data from the titration period and M1 will not be used for evaluation of the confirmatory endpoints. Consequently, it was essential to extend the trial duration in order to accommodate M2 (36 weeks) during which the confirmatory endpoints related to hypoglycemia will be assessed. This will allow sufficient data collection for evaluation of the scientific objectives while preserving the integrity of the primary endpoint. In keeping with the same principles, the confirmatory test hierarchy was modified to exclude the endpoint of severe or BG-confirmed symptomatic hypoglycemia during the total treatment period which will be analyzed as a secondary endpoint instead. Empirical evidence to confirm the conclusions from the primary analysis will be obtained via relevant sensitivity analyses. As indicated in Table 4, two sensitivity analyses are prespecified exclusively to address the impact of the protocol amendment on the primary endpoint. These considerations will ensure robust evaluation of the confirmatory endpoints.
At the time of the amendment, all patients had good glycemic control after 16 weeks of titration (Figure 2). Since there was no change in analysis period or definition of the primary endpoint, recalculation of the sample size or introduction of a new titration period was not deemed necessary. In addition, a high retention rate was observed during the trial leading to a sufficiently high power (~79%) on the primary endpoint at the time of the amendment. Therefore, it was considered reasonable to continue the trial without recruiting additional patients.
All necessary measures were directed toward ensuring patient safety and maintaining scientific trial integrity following the postinitiation amendment. Importantly, the investigators and trial site personnel are committed to providing optimal treatment and care for their patients, while minimizing the impact of the amendment. This is also reflected by the fact that the majority of patients on-treatment at the time of amendment reconsented to continue their participation with ~1% withdrawing due to the amendment. Importantly, other than the concerns regarding the glycemic data collection system, no unexpected adverse events have been reported by patients, thus far.
Conclusion
In order to discern the effect of long-acting basal insulins, degludec and glargine U300, on hypoglycemia, a randomized controlled trial is under way. The postinitiation protocol amendment for this trial was mandated due to unusual reporting of glycemic parameters and hypoglycemia data owing to the glycemic data collection system. Importantly, critical safety concerns identified in this trial were addressed in a timely manner ensuring that trial participants were not subject to untoward risks. The amended trial design will ensure that the trial outcomes can be evaluated using a rigorous scientific method. The observations from this trial underscore the importance of ongoing medical monitoring and surveillance activities during trial conduct. In addition, our experience provides an insight into handling major safety-related amendments during ongoing trials that could be applicable to future studies.
Footnotes
Acknowledgements
The authors would like to thank Deniz Tutkunkardas, MD, Novo Nordisk A/S, for scientific review of the manuscript and Ruchita Kapoor, PhD, Novo Nordisk A/S, for medical writing support.
Abbreviations
ANCOVA, analysis of covariance; BG, blood glucose; BGM, blood glucose meter; CE, Conformité Européenne; degludec, insulin degludec; FPG, fasting plasma glucose; FU, follow-up visit; glargine U100, insulin glargine 100 units/mL; glargine U300, insulin glargine 300 units/mL; ITT, intent-to-treat; M1, maintenance period 1; M2, maintenance period 2; MGH, MyGlucoHealth; MMRM, mixed model repeated measures; OAD, oral antidiabetic drug; OD, once daily; SMBG, self-measured blood glucose; T2D, type 2 diabetes.
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: APT is employed by Scripps Health, which provides research and consultancy services to Novo Nordisk A/S. IS has received payment for consultancy services to Novo Nordisk A/S. LNT and BAB are employees and shareholders of Novo Nordisk A/S. LAL has received research funding from, has provided CME on behalf of, and/or has acted as an advisor to AstraZeneca, Boehringer Ingelheim, Eli Lilly, GSK, Janssen, Merck, Novo Nordisk, Sanofi, and Servier.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Novo Nordisk A/S.