
Abstract
Background
Lung adenocarcinoma (LUAD) stands as a major respiratory malignancy with high mortality. With the advent of immunotherapy, new therapeutic avenues have emerged in cancer treatment.
Objective
Our focus aimed at developing a CD8+ T cell-based immune gene prognostic model (CDIGPM) for LUAD, shedding light on the immunological aspects and the potential advantages of immunotherapy in distinct CDIGPM-defined LUAD categories.
Methods
Data from LUAD patients were extracted from the TCGA and GEO databases (GSE11969). The differentially expressed genes (DEGs) were intersected with immune genes from ImmPort and InnateDB, yielding 89 significant immune genes related to CD8+ T cells (CDIGs). Univariate Cox regression and LASSO regression analyses were performed on 10 hub CDIGs (ADM, CAV1, CTSL, HLA-DMB, HLA-DQA1, IGHM, PLSCR1, PTGDS, S100A16, and WFDC2). Furthermore, the immunological attributes and the immunotherapy efficacy in CDIGPM-defined categories were explored. Moreover, to support the findings of the bioinformatics analysis, fifteen LUAD patients’ tumor and adjacent tissues were collected for qRT-PCR detection of CDIGPM-related genes.
Results
Kaplan-Meier analysis revealed that the high-CDIGPM group exhibited significantly poorer overall survival (OS) trajectories, whereas the low-CDIGPM group showed more favorable OS trajectories, indicating a better prognosis. Age, tumor stage, and CDIGPM score were identified as independent prognostic factors. The high-CDIGPM group was enriched in pathways related to the cell cycle, focal adhesion, and cancer, while the low-CDIGPM group was associated with immune response-related pathways. The CDIGPM model effectively differentiated clinical subtypes in patients with LUAD. QRT-PCR detection of Clinical LUAD samples also validated the differentially expression of CDIGPM model related genes.
Conclusions
The study highlights the prognostic importance of CDIGs in LUAD using the CDIGPM model, linking age, stage and CDIGPM score to poor outcomes. The identified genes and pathways provide potential therapeutic targets, deepening our understanding of LUAD's molecular landscape.
Introduction
Lung adenocarcinoma (LUAD), a major subtype of non-small cell lung cancer (NSCLC), remains the leading global cause of cancer-related deaths, accounting for nearly half of all cases. 1 Despite therapeutic advancements, the 5-year survival rate for LUAD remains suboptimal at under 20%.2–4 The introduction of immunotherapies targeting immune checkpoints has revolutionized the treatment of LUAD. 5 While biomarkers like PD-L1 expression and tumor mutation burden (TMB) are clinically utilized to predict immunotherapy outcomes, they don't fully capture the diversity of the tumor microenvironment (TME).6,7 Hence, the development of predictive models and the discovery of novel biomarkers for prognosis and treatment efficacy is imperative.
The TME, a multifaceted milieu consisting of various immune cells, stromal cells, and cytokines, significantly influences tumor development and progression, with adaptive immune responses carried out by immune cells being crucial. 8 Alterations in the TME can impact patient prognosis and serve as potential biomarkers for immunotherapy. 9 However, the presence of numerous immunosuppressive cells or chemicals in the TME impairs the function of CD8+ T cells, which are the TME's principal antitumor effector cells and primarily serve a cytotoxic role. Additionally, as the immune response slows, co-inhibitory molecules such as programmed death protein 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4) are increased on the surface of CD8+ T cells, leading to T cell exhaustion in the TME. Understanding the regulation of CD8+ T cells in the TME is crucial.
The advent of single-cell RNA-sequencing (scRNA-seq) provides a unique perspective for exploring the molecular nuances of diverse immune cells within the TME. Leveraging scRNA-seq data to explore gene expression patterns linked to immune cell characteristics can aid in predicting cancer prognosis and outcomes of immunotherapy. In Zhang's study, scRNA-seq was used to thoroughly analyze LUAD, emphasizing the molecular traits of tumor-infiltrating CD8+ T cells and identifying specific markers for these CD8+ T cells. 10 Chen et al. utilized both scRNA-seq and bulk RNA-seq to study the TME and intra-tumor heterogeneity at various stages of LUAD, examining the single-cell expression patterns of alveolar cells, early-stage malignant cells, and advanced-stage malignant cells. The cellular diversity and molecular complexity of immune cell lineages at various stages of LUAD were also investigated. 11
Therefore, this study aimed to identify CD8+ T cell-associated marker genes to build a multi-gene signature and design a predictive risk model for LUAD using immune cell line-associated information. The findings will enhance prognosis and predictions of immunotherapeutic response in LUAD patients, thereby improving treatment outcomes.
Materials and methods
Data acquisition and initial processing
We collected 210 CD8+ T-cell-associated differentially expressed genes (DEGs) from the Tumor Immune Single Cell Hub (TISCH) database (http://tisch1.comp-genomics.org/home/), 12 drawing from the GSE11969 dataset specific to LUAD single cells. Additionally, 2,533 genes linked to the immune system were identified from both the InnateDB (https://www.innatedb.ca/index.jsp) and ImmPort (https://www.immport.org/home) datasets.13,14 Clinical, mutational, and LUAD RNA-seq data were extracted from the TCGA database via the GDC Data Portal (https://portal.gdc.cancer.gov/). 15 By applying a threshold of P < 0.05 and an absolute log2 fold change (FC) of ≥1.5, the DEGs were cross-referenced with the immune genes from the aforementioned sources, yielding 89 immune genes related to CD8+ T cells (CDIGs) (Figure 1(a)).

Recognition of hub CD8+ T cell-associated immune genes (CDIGs). (a) The overlapped genes of differentially expressed genes (DEGs) obtained from the Tumor Immune Single Cell Hub (TISCH) database in CD8+ T cells in lung adenocarcinoma (LUAD) single-cell dataset GSE11969 and immune-related genes acquired from the InnateDB and ImmPort datasets. (b) The PPI network of CDIGs. (c) CDIGs screened by the number of adjacent nodes ≥20.
Analysis of protein-protein interaction (PPI)
The STRING database was used to conduct PPI analysis, identifying key CDIGs with at least 20 adjacent nodes.
CDIGPM development
Survival analysis revealed significant variations in 27 out of the 89 genes, identifying ten genes through both univariate and multivariate Cox regression analysis. The efficacy of the resulting CDIGPM was assessed using Kaplan-Meier survival curves and a log-rank test on both the TCGA and the GSE11969 cohort from GEO. Additionally, Receiver Operating Characteristic (ROC) curves were generated for the TCGA cohort to assess the predictive capacities of various factors.
GSEA and analysis of somatic mutations
GSEA software (version 4.1.0) 16 was utilized to assess whether significant differences existed in gene expression between the CDIGPM low and high groups, focusing on the enrichment within the MSigDB Collection. Somatic mutation data for LUAD samples, formatted as “maf”, were sourced from the TCGA GDC Data Portal. The “Maftools” package in R software (version 4.2.3; https://www.r-project.org/) 17 facilitated the creation of waterfall plots for visualizing and summarizing the mutations in the genes.
Examining immune landscape and clinical subgroups related to CDIGPM
Correlation analyses were executed to understand the connection between CDIGPM and CD274 expressions. The expression data for LUAD samples were input into CIBERSORT (https://cibersort.stanford.edu/) 12 and processed 1000 times to estimate the proportions of 22 immune cell types. The percentages of these cell types were then compared between the two CDIGPM subgroups, with the findings illustrated on a landscape map. The heat map and circle chart provide a visual representation of the CDIGPM groups and clinical subtypes for LUAD patients from the TCGA cohort, including age, gender, stage, and TNM stage.
qRT-PCR validation
To support the findings of the bioinformatics analysis, fifteen LUAD patients weer enrolled, tumor and adjacent tissues were collected. All the patients were informed and signed, and this study was approved by the Ethics Committee of Tongde Hospital of Zhejiang Province (Approve no. 2024095-JY). The clinical samples were used for qRT-PCR validation of CDIGPM-related genes. Briefly, total RNA was extracted from tumor and adjacent tissue samples, DNA contamination was removed using DNase treatment. Next, RNA was converted into cDNA by reverse transcription reaction. cDNA was then amplified by designing specific primers and using qRT-PCR instrument while detecting fluorescence signals. Finally, the relative expression of the target gene was calculated by comparing the Ct value and combining with the internal reference gene. The primer sequences of the genes were list in Table 1.
Primer sequence of genes.

Statistical analysis
For the analysis of continuous variables, an independent samples t-test was used; for categorical data, the χ² test was used. The log-rank test and Kaplan-Meier survival analysis were utilized in the univariate survival study to evaluate the differences in survival rates among groups. The multivariate survival study was conducted using the Cox regression model to analyze the impact of multiple variables on survival outcomes. Significant differences were indicated by a P value of less than 0.05.
Results
CDIGs identification
TISCH provided the 210 DEGs in CD8+ T cells in LUAD. The investigation of the tumor microenvironment (TME) is facilitated by TISCH, a scRNAseq database that focuses on the TME. P < 0.05 and | log2 FC| ≥ 1.5 were used as the thresholds for DEG filtering in CD8+ T cells. In the meantime, ImmPort and InnateDB were utilized to obtain the most up-to-date information on immune genes. Later, 89 CDIGs were generated by intersecting immunological genes and DEGs in CD8+ T cells (Figure 1(a)). These CDIGs formed a complex PPI network, with nine CDIGs having a higher adjacent node (adjacent node count > 20) (Figure 1(b) and (c)).
CDIGPM model construction
Univariate Cox regression analysis identified 27 out of the 89 genes (CDIGs) with significant relevance to overall survival (OS) (Figure 2(a)), indicating their potential role in patient prognosis. In Figure 2(b), LASSO regression was employed to assess the influence of these 27 prognostic genes. The coefficient profiles derived from this analysis elucidate the relative importance of each gene in the prognostic model. In Figure 2(c), utilizing tenfold cross-validation in the LASSO regression model, a subset of 10 genes was identified as the most significant predictors. In Figure 2(d), Kaplan-Meier curves of 10 hub CDIGs (ADM, CAV1, CTSL, HLA-DMB, HLA-DQA1, IGHM, PLSCR1, PTGDS, S100A16, and WFDC2) in the TCGA cohort were presented. These genes were selected based on the minimum lambda value, which ensures optimal model performance while minimizing overfitting in the CDIGPM model.
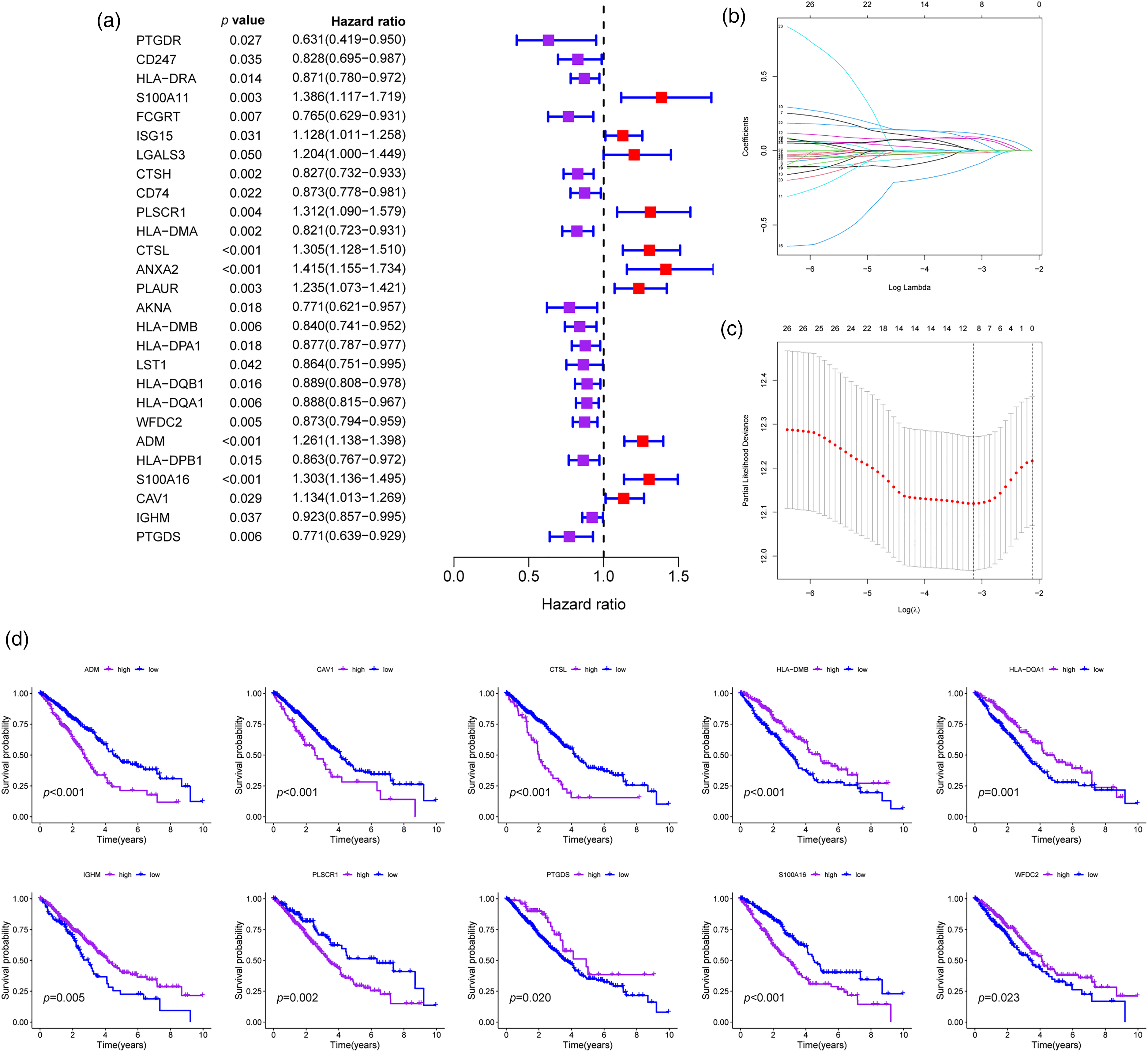
CD8+ T cell-associated immune gene prognostic model (CDIGPM). (a) 27 out of the 89 hub CDIGs show remarkable relevance to overall survival (OS) according to univariate Cox regression analysis. (b) LASSO coefficient profiles of 27 prognostic genes. (C) LASSO regression with tenfold cross-validation obtained 10 prognostic genes using minimum lambda value. (d) Kaplan-Meier survival analysis of 10 hub CDIGs (ADM, CAV1, CTSL, HLA-DMB, HLA-DQA1, IGHM, PLSCR1, PTGDS, S100A16, WFDC2) in the TCGA cohort.
CDIGPM elucidates significant survival implications on LUAD patients
In Figure 2(d) and (e), the Kaplan-Meier survival analysis on both TCGA and GEO cohorts revealed distinct survival curves for high- and low-CDIGPM groups. The high-CDIGPM group showed lower OS trajectories, indicating poorer prognosis, while the low-CDIGPM group displayed higher OS trajectories, signifying favorable prognosis in both datasets. We further examined the correlation between individual gene expression within the CDIGPM and OS in patients with LUAD. As illustrated in Figure 3, elevated expression levels of ADM, CAV1, CTSL, PLSCR1, and S100A16 were significantly associated with reduced survival times in LUAD patients compared to those with lower expression levels of these genes. Conversely, diminished expression of HLA-DMB, HLA-DQA1, IGHM, PTGDS, and WFDC2 was significantly linked to poorer prognosis in LUAD patients.

Kaplan-Meier curves of high- and low-CDIGPM groups. (a) Kaplan-Meier survival analysis of high- and low-CDIGPM groups in the TCGA cohort. (b) Kaplan-Meier survival analysis of the high- and low-CDIGPM groups in the GEO cohort.
Using univariate and multivariate Cox regression analysis, we then assessed the independent prognostic significance of CDIGPM. Results indicated that age, tumor stage, and CDIGPM score were independent predictive variables (P < 0.05) (Figure 4(a) and (b)). In Figure 4(c), the prognostic efficacy of the CDIGPM was further confirmed by evaluating the area under the curve (AUC) of ROC curve in the TCGA cohort. The results indicated that, when controlling for age, gender, and tumor stage, only the AUC value of the risk score group exceeded 0.7. This finding suggests that the CDIGPM model developed in this study demonstrates strong diagnostic capability.
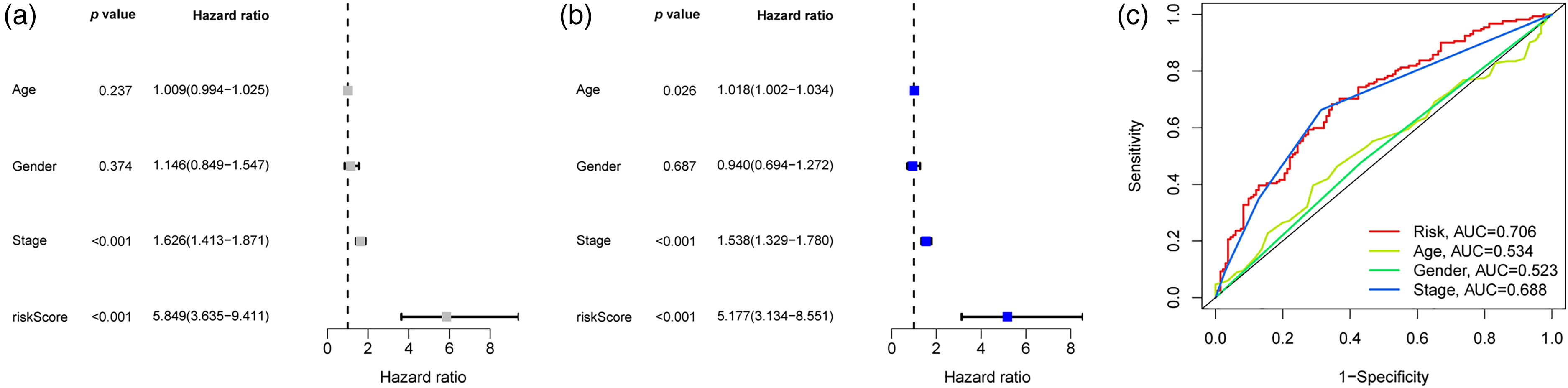
Evaluated the independent prognostic value of CDIGPM via univariate and multivariate Cox regression analyses. (a) Univariate Cox analysis of clinical factors and the CDIGPM. (b) Multivariate Cox analysis of the factors significant in the univariate Cox analysis. (c) The ROC curves of the risk score, age, gender, grade, and stage in the TCGA cohort.
GSEA and somatic mutations Analysis in high- and low- CDIGPM groups
The two CDIGPM groups’ enriched gene sets for KEGG pathway were identified using GSEA, with the top five routes plotted. The findings revealed that the high-CDIGPM group had enriched levels of the cell cycle, focal adhesion, cancer-related pathway, actin cytoskeleton regulation, and spliceosome (Figure 5(a)). Additionally, asthma, the intestinal immune network for IgA synthesis, systemic lupus erythematosus, and immune response-related pathways (such as allograft rejection and alpha linolenic acid metabolism) were also enriched in low-CDIGPM group (Figure 5(b)). Subsequently, gene mutations were analyzed and illustrated in Figure 5(c) and (d) for low and high CDIGPM groups.

GSEA and mutation in high- and low-CDIGPM groups. (a) KEGG pathway-related gene sets are enriched in the high-CDIGPM group. (b) KEGG pathway-related gene sets are enriched in the low-CDIGPM group. (c, d) Significantly mutated genes in the mutated LUAD samples were in high-CDIGPM group (c) and low-CDIGPM group (d). Mutated genes (rows) are ordered by mutation rate; samples (columns) are arranged to emphasize mutual exclusivity among mutations. The right shows mutation percentage, and the top shows the overall number of mutations. The color-coding indicates the mutation type.
Characteristics of the two CDIGPM groups’ immune systems
The relationship between the CDIGPM score and immune checkpoint genes was then investigated to determine any potential correlations. The necessity of choosing CD274 (also known as PD-L1) instead of other immune checkpoints lies in its unique role in the immune response and its potential for therapeutic targeting. The CDIGPM score consequently showed a favorable correlation with CD274 expression (Figure 6(a) and (b)). The two CDIGPM groups displayed distinct immune cell compositions. In the low-CDIGPM group, more follicular helper T cells, resting dendritic cells, resting mast cells, and eosinophils were found. In contrast, the high-CDIGPM group had more activated memory CD4+ T cells, resting NK cells, resting macrophages M0, active mast cells, and activated neutrophils (Figure 7(a) and (b)). Then, using specific gene signatures, we distinguished the immunological and molecular functions between the two groups. As a result, the low-CDIGPM group's immunological and molecular functions were more active (Figure 7(c)).

The expression of CD274 in high- and low-CDIGPM groups. (a) CD274 expression in high- and low-CDIGPM groups. (b) Correlation analysis between CDIGPM and CD274 expression.
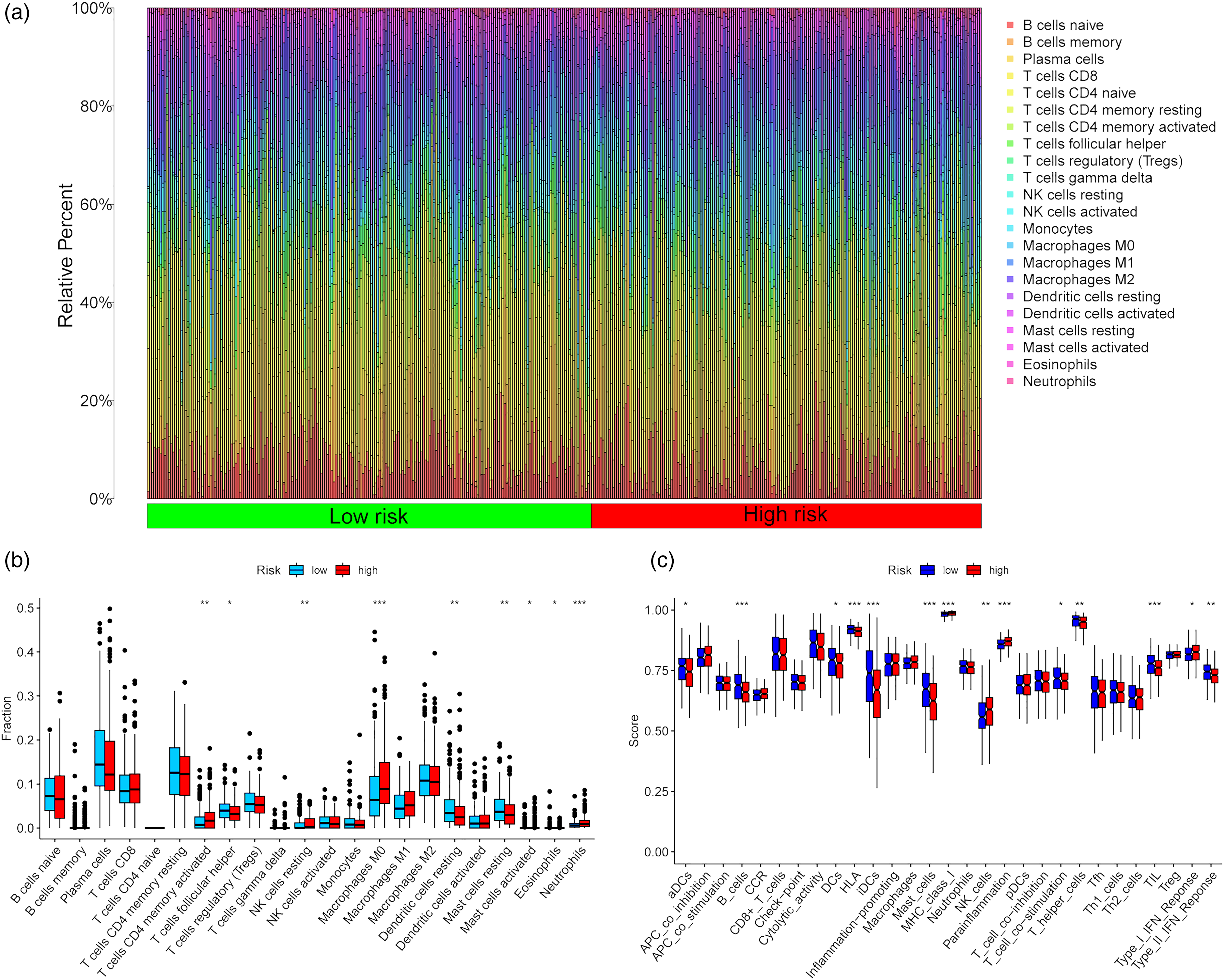
Immune characteristics in high- and low-CDIGPM groups. (a, b) The proportions of tumor microenvironment (TME) cells in high- and low-CDIGPM groups. (c) The molecular and immune-related function in high- and low-CDIGPM groups (*p < 0.05, **p < 0.01, ***p < 0.001).
Interaction between CDIGPM and Clinical Subtypes in LUAD Patients
Figure 8(a) and (b) displays a strong association between high- and low-CDIGPM risk groups and various clinical subtypes for LUAD patients in the TCGA cohort, with significant correlations observed with gender, stage, TNM (T and N) stage, but not age. The low-risk group had a higher proportion of females and lower stages. Figure 8(c) shows significant differences in immune subtypes (C1-C4, C6) between high- and low-CD8 + T cell groups, highlighting the influence of CD8+ T cell abundance on the distribution of immune subtypes in LUAD patients.
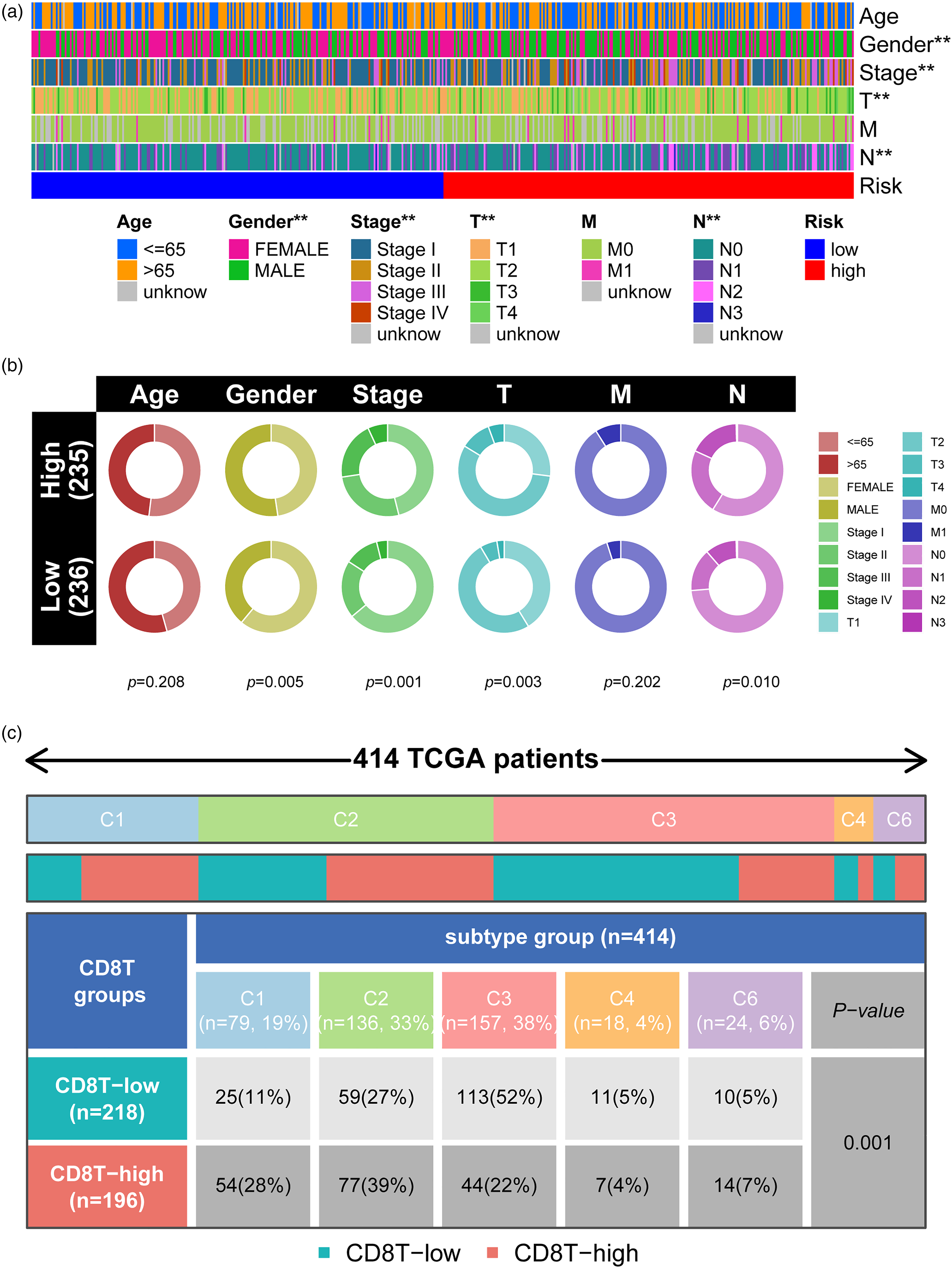
Relationship between CDIGPM and clinical subtypes. (a-b) Heat map and circle chart of the CDIGPM groups and clinical subtypes for LUAD patients in the TCGA cohort. Age, gender, stage, and TNM stage are shown as patient annotations. (c) Comparison of the differences in immune subtype (C1-C4, C6) between high- and low-CD8+ T groups. **p < 0.01.
Validation of CDIGPM-related genes expression in clinical LUAD patients
As shown in Figure 9, the elevated expression levels of ADM, CAV1, CTSL, PLSCR1, and S100A16 were observed in LUAD patient tumor tissues compared to those in adjacent tissues. Conversely, decreased expression levels of HLA-DMB, HLA-DQA1, IGHM, PTGDS, and WFDC2 were found in LUAD patients tumor tissues compared to those in adjacent tissues.

QRT-PCR detection of ten CDIGPM-related genes in clinical LUAD patients’ tumor and adjacent tissues. n = 15, compared to the adjacent tissues, ▴p < 0.05, ▴▴p < 0.01.
Discussion
Immunotherapy stands out as a leading treatment option for various tumors, including LUAD. This treatment modality functions by stimulating autologous immune reactions, disrupting the body's tolerance to cancer, and revitalizing mechanisms that resist tumors within the TME.18–20 Nevertheless, the intricate mechanisms that underpin tumor immunity led to unsatisfactory results for some patients. 21 The landscape of LUAD treatment has witnessed progress with the emergence of immunotherapies, notably anti-CTLA4 and anti-PD-1 antibodies. 22 Although anti-PD-1 antibody therapy may yield sustained responses, resistance still occurs in around 60% of patients. Several factors, including cancer cell phenotype plasticity, alterations in the tumor microenvironment, and the expression levels of immune checkpoint genes, may play a role in LUAD's immune escape. 23 Moreover, the precision of existing genomic and immune biomarkers in assessing the efficacy of therapies is suboptimal. 24 Consequently, identifying more reliable predictors is imperative for the precise evaluation of clinical outcomes prior to the administration of immunotherapy.
T cells serve as crucial tumor-resistant effector cells due to their direct engagement with and assault on cancer cells. 25 Recent findings have highlighted that immune checkpoint therapy (ICT) aimed at T cells yields promising results in LUAD instances. However, the effectiveness of ICT is selective, proving beneficial only in specific tumor scenarios. This selectivity in efficacy appears to be influenced by the degree of activation and the presence of infiltrating immune cells, particularly CD8+ T lymphocytes.26,27 In our study, the identification of 210 DEGs in CD8+ T cells from the TISCH database underscores the importance of scRNA-seq in understanding the TME. The intersection of these DEGs with immune genes from ImmPort and InnateDB to obtain CDIGs further refines the list of genes that are crucial in the TME. Univariate Cox regression and LASSO regression pinpointed 10 of CDIGs (ADM, CAV1, CTSL, HLA-DMB, HLA-DQA1, IGHM, PLSCR1, PTGDS, S100A16, and WFDC2) as the most significant predictors, forming the basis of our CDIGPM model. QRT-PCR validation based on clinical LUAD patients also found differentially expression of these CDIGPM model related genes between tumor and adjacent tissues, which also implies the reliability of the model. Kaplan-Meier survival analysis on both TCGA and GEO cohorts showcased the model's robustness in predicting survival outcomes. Similarly, for model construction, in Liu et al.'s 28 study, they also applied univariate and multivariate Cox regression analyses, as well as LASSO Cox analysis with ten-fold cross-validation to construct a immune-related 13-lncRNA signature scoring model to predict disease-free survival of endometrial carcinoma. Mao et al 29 also used this method to construct a risk score model based on the lncRNAs associated with ferroptosis subtypes in LUAD. Zhang et al. 10 identified 252 genes related to CD8+ T cells and two LUAD subtypes (IC1 and IC2) using a CD8+ T cell-associated gene signature. IC2 patients had higher tumor mutation rates and lower immune responses, while IC1 patients demonstrated better responses to immune checkpoint inhibitors. The correlation analyses between CDIGPM and CD274 expressions reveal significant insights into the immune landscape of the studied samples. CD8+ T cells play a critical role in anti-tumor immunity, and their activity is often modulated by immune checkpoint molecules such as CD274 (PD-L1). By investigating the relationship between CDIGPM and CD274 expressions, we can infer how the immune gene signatures associated with CD8+ T cells might influence or be influenced by the presence of CD274. A strong positive correlation between CDIGPM and CD274 expression may indicate that tumors with higher infiltration of CD8+ T cells also express elevated levels of CD274. This could suggest an adaptive resistance mechanism, where tumors upregulate CD274 in response to immune pressure exerted by CD8+ T cells. Such findings would underscore the importance of targeting both CD8+ T cell activation and immune checkpoints in therapeutic strategies. Using top genes, they created a 10-gene signature, validated with GSE and TCGA datasets, demonstrating reliable prognosis and immunotherapy response prediction in LUAD patients. 10
Molecular analysis revealed distinct pathways enriched in high and low CDIGPM groups, with the former showing a predilection for cell cycle and cancer pathways, and the latter leaning towards immune responses. Immune cell composition varied between the two groups, with the low-CDIGPM group exhibiting more active immune and molecular functions. Indicating the low CDIGPM group exerted the anti-tumor effect through the immune response. This suggests that the low CDIGPM group is more adept at mounting an anti-tumor immune response, which is crucial for controlling and eliminating cancer cells. For instance, a study found that enhanced cell cycle activity (CCA) is often associated with unfavorable clinical outcomes across various cancer types. 30 However, cancer cohorts treated with immune checkpoint blockade therapy did not show this relationship, indicating a complex interplay between cell cycle activity and anti-tumor immunity. Another study discovered that Glioma stem cells (GSCs) were more susceptible to interferon-beta (IFN-β) inhibition than normal stem cells (NSCs). According to the genomic analysis, immune response and cell adhesion genes were upregulated whereas the cell cycle and ribosome pathways were downregulated, suggesting a balance between immune response and cell cycle regulation in the anti-tumor effect of IFN-β.31,32 These findings underscore the importance of understanding the distinct molecular and immune profiles of high and low CDIGPM groups. Recognizing the pathways enriched in each group can provide valuable insights into the mechanisms driving their anti-tumor effects, or lack thereof. This knowledge is crucial for developing targeted therapeutic strategies that can effectively harness the immune system's power to fight cancer while considering the molecular characteristics of the tumor.
Finally, a comprehensive association between CDIGPM scores and clinical subtypes in LUAD patients was depicted, emphasizing the potential influence of CD8+ T cell abundance on immune subtype distribution. This study underscores the importance of CDIGPM in understanding the TME of LUAD and its potential as prognostic marker. To further elaborate, a study discussed the dynamic process of CD8+ T cell differentiation and functional exhaustion. 33 It also identifies potential biomarkers in this process, providing valuable targets for therapy and immunotherapy in LUAD patients. 33 This study supports the notion that understanding the behavior and characteristics of CD8+ T cells in LUAD is crucial for developing effective therapeutic strategies and improving patient outcomes. Another study found that two ferroptosis subtypes can predict clinical outcomes and therapeutic responses in patients with LUAD. 29 This finding suggests that different molecular and genetic markers, including those related to CD8+ T cells and ferroptosis, can provide valuable information for predicting patient outcomes and personalizing treatment plans in LUAD. 29 Furthermore, the study highlights the differences in characteristics between the two LUAD subtypes, which can enhance current detection and treatment methods. The study identifies a transcription factor, BACH1, that is significantly upregulated in one of the tumor subtypes, suggesting that the pathways activated by this factor may be associated with tumor progression and poor prognoses. 34 In light of these findings, it is evident that the abundance and characteristics of CD8+ T cells, along with other molecular and genetic markers, play a significant role in the prognosis and treatment of LUAD. The CDIGPM scores, as indicated in the initial study, serve as an important tool for understanding the complex landscape of LUAD, providing clinicians and researchers with valuable insights for developing more effective and personalized therapeutic approaches for patients suffering from this type of cancer.
In conclusion, a CD8+ T cell-based immune gene prognostic model CDIGPM was developed, and found that this model was associated with OS trajectories and effectively differentiated clinical subtypes in patients with LUAD. This indicated that the CDIGPM model serves as a promising tool for understanding the TME of LUAD patients, predicting patient prognosis, and guiding therapeutic interventions. The interplay between CDIGs, immune cells, and clinical subtypes underscores the complexity of LUAD and highlights the need for personalized medicine approaches. As this research is the result of bioinformatics analysis, further clinical studies are warranted to validate these findings in larger cohorts and to confirm the accuracy of the CDIGPM model and explore the therapeutic potential of the identified CDIGs.
Footnotes
Acknowledgements
Not applicable.
Ethical statement
This study was approved by the Ethics Committee of Tongde Hospital of Zhejiang Province (Approve no. 2024095-JY), all the patients were informed and signed.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Authors’ contributions
CONCEPTION: Keke Tang and Hainan Ye; INTERPRETATION OR ANALYSIS OF DATA: Jia Che and Dan Liu; PREPARATION OF THE MANUSCRIPT: Keke Tang, Qifen Mao, Yixuan Zhou; REVISION FOR IMPORTANT INTELLECTUAL CONTENT: Ji Shen; SUPERVISION: Hainan Ye.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Zhejiang Medical Health Science and Technology Project under Grant Keke Tang [2023KY285].
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request