
Abstract
Background:
Radiographic axial spondyloarthritis (r-axSpA), formerly known as ankylosing spondylitis (AS), is a chronic, inflammatory rheumatic disease associated with symptoms such as inflammatory back pain, morning stiffness, and arthritis. First-line recommendations for patients with AS include treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) for reducing pain and stiffness.
Objectives:
The objective of our study is to evaluate the efficacy and short-term NSAID-sparing effect of secukinumab in patients with AS currently treated with NSAIDs.
Design:
We assessed the clinical Assessment of SpondyloArthritis International Society (ASAS20) response to secukinumab and evaluated the extent to which the use of concomitant NSAID can be reduced between weeks 4 and 12 in r-axSpA patients treated with secukinumab 150 mg compared with placebo.
Methods:
ASTRUM was a prospective 24-week randomized controlled trial of adult patients with active r-axSpA [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ⩾4] who had a documented inadequate response to ⩾2 NSAIDs. Patients were randomized (1:1:1) to initiate treatment with subcutaneous secukinumab 150 mg from either week 0 (group 1), week 4 (group 2), or week 16 (group 3). From week 4 onward, tapering of NSAIDs was allowed in all groups.
Results:
This study included 211 patients (n = 71, 70, and 70 in groups 1, 2, and 3, respectively). ASAS20 response at week 12 for pooled groups 1 and 2 versus group 3 was 51.1% versus 44.3% (p = 0.35). A higher proportion of patients in groups 1 and 2 achieved ASAS40 and BASDAI50 and showed improvements in other secondary clinical outcomes as compared to group 3 at week 16. More patients in groups 1 and 2 versus group 3 stopped their NSAID intake from baseline through week 16.
Conclusion:
Treatment with secukinumab improved clinical outcomes and showed a short-term NSAID-sparing effect in patients with r-axSpA, even though the primary endpoint was not met.
Trial registration:
ClinicalTrials.gov; NCT02763046, EudraCT 2015-004575-74.
Introduction
Radiographic axial spondyloarthritis (r-axSpA), formerly known as ankylosing spondylitis (AS), is a chronic, immune-mediated, inflammatory rheumatic disease associated with symptoms such as inflammatory back pain, morning stiffness, anterior uveitis, enthesitis, and arthritis, which may lead to impaired mobility and joint function, and a reduced health-related quality of life (HR-QoL).1–3 It primarily affects the axial skeleton, including the sacroiliac joints and the spine, but also extra-musculoskeletal organs such as the skin, the gut, the eye, and the heart.4,5
Based on the European Alliance of Associations for Rheumatology (EULAR) and the American College of Rheumatology (ACR) guidelines for patients with AS, the first-line recommendations include treatment with nonsteroidal anti-inflammatory drugs (NSAIDs) and physical therapy, while biologic-disease-modifying antirheumatic drugs (bDMARDs) including tumor necrosis factor inhibitors (TNFi) and interleukin-17 inhibitors (IL-17i) are considered as the second-line treatment for reducing pain and stiffness.6–9 Evidence from recent studies demonstrates inhibition of the Janus kinase (JAK) pathway with targeted synthetic (ts)DMARDs to be a good additional therapeutic option for managing r-axSpA.10–12 Furthermore, bimekizumab, a dual IL-17A and IL-17F inhibitor, has also demonstrated sustained improvement in patients with active r-axSpA. 13
In Germany, results from a claims data analysis showed that the prescription frequency of NSAIDs is about 50% in patients with r-axSpA. 14 However, there is a potential risk of toxic side effects on gastrointestinal, renal, and cardiovascular systems due to prolonged use of NSAIDs in patients with r-axSpA.15–22 Therefore, it is important that the long-term safety of NSAIDs should be assessed to evaluate the feasibility of an overall dose reduction of NSAIDs, which does not lead to increased pain levels in patients with r-axSpA.
According to international recommendations by ACR/EULAR, use of bDMARDs such as TNFi and IL-17A inhibitors should be considered for controlling the symptoms in patients with r-axSpA who do not respond to or have contraindications to NSAIDs. 6 Concomitantly with NSAIDs, bDMARDs are initially often prescribed in clinical practice. Several clinical trials have reported that TNFi may lead to successful NSAID tapering in r-axSpA patients.5,23,24 However, currently, there are no clear recommendations for tapering or discontinuing NSAIDs after the initiation of bDMARDs. Previous trials on the NSAID-sparing effect of etanercept in r-axSpA reported that a substantial reduction in NSAID intake was associated with lower ASAS20 response rates (44% versus 57% at week 8) compared to phase III clinical data.17,25 Results from a recent post hoc analysis of pooled data from the MEASURE 2–4 studies showed an increase in the proportion of patients achieving a 50% reduction in the ASAS-NSAID score and very low ASAS-NSAID scores of <10 (clinically meaningful reduction in NSAID intake) over time in both low and high NSAID intake patients. Similarly, the percentage of patients with a clinically relevant improvement [Bath Ankylosing Spondylitis Disease Activity Index (BASDAI) ⩽ 2] was consistently higher in patients with an ASAS-NSAID score < 10 as compared to those with ASAS-NSAID score ⩾ 10. 26 Two questions emerge based on these findings: (i) what is the extent to which the use of concomitant NSAID can be reduced and (ii) what is the optimal time point to initiate tapering of NSAID. To answer these questions, the ASTRUM (NCT02763046) study was planned to examine the efficacy and NSAID-sparing effect in secukinumab-treated patients with r-axSpA over a period of up to 24 weeks.
Secukinumab, a human monoclonal antibody that directly inhibits IL-17A, has demonstrated sustained efficacy and a consistent safety profile in patients with r-axSpA.27–33 The current study assessed the clinical ASAS20 response to secukinumab and evaluated the extent to which the use of concomitant NSAID can be reduced between weeks 4 and 12 in r-axSpA patients treated with secukinumab 150 mg compared with placebo.
Materials and methods
Study design
The ASTRUM study, conducted across 41 sites in Germany, was a 24-week, randomized, double-blind, 3-arm, placebo-controlled, parallel-group, phase IV study of secukinumab in patients with r-axSpA. To be considered TNF inhibitor inadequate responders (TNF-IR), patients treated with a maximum of two TNFi must have shown an insufficient response to TNFi for more than 3 months or shown intolerance to at least one dosage of one TNFi. Patients were stratified randomly according to prior TNFi exposure, that is, TNFi-naïve patients or TNF-IR, with a minimum of 60% TNFi-naïve patients and a maximum of 40% TNF-IR patients.
The enrolled patients were randomized (1:1:1) to one of the three treatment groups based on the initiation of secukinumab treatment: group 1 received subcutaneous (s.c.) secukinumab 150 mg at baseline, weeks 1, 2, 3, and 4 and then every 4 weeks until week 20; with placebo injections at weeks 5, 6, 7 and 17, 18, 19 to maintain blind; group 2 received weekly secukinumab 150 mg s.c. from week 4 until week 8 and then once in every 4 weeks until week 20, where they received placebo every week from baseline until week 4 to maintain blind; group 3 patients received weekly dose of placebo from baseline to week 8 and then at weeks 12 and 16, but from week 16, they received weekly dose of secukinumab 150 mg s.c. until week 20 (Figure 1). Patients were told to report their daily intake of NSAID and the level of AS neck, back, and hip pain daily in a paper-based patient diary. There were no restrictions on patients to take any commercially available NSAIDs in Germany. In all three groups, tapering of NSAIDs was allowed from week 4 onward. The decision was based on clinical information without a prespecified reduction scheme.
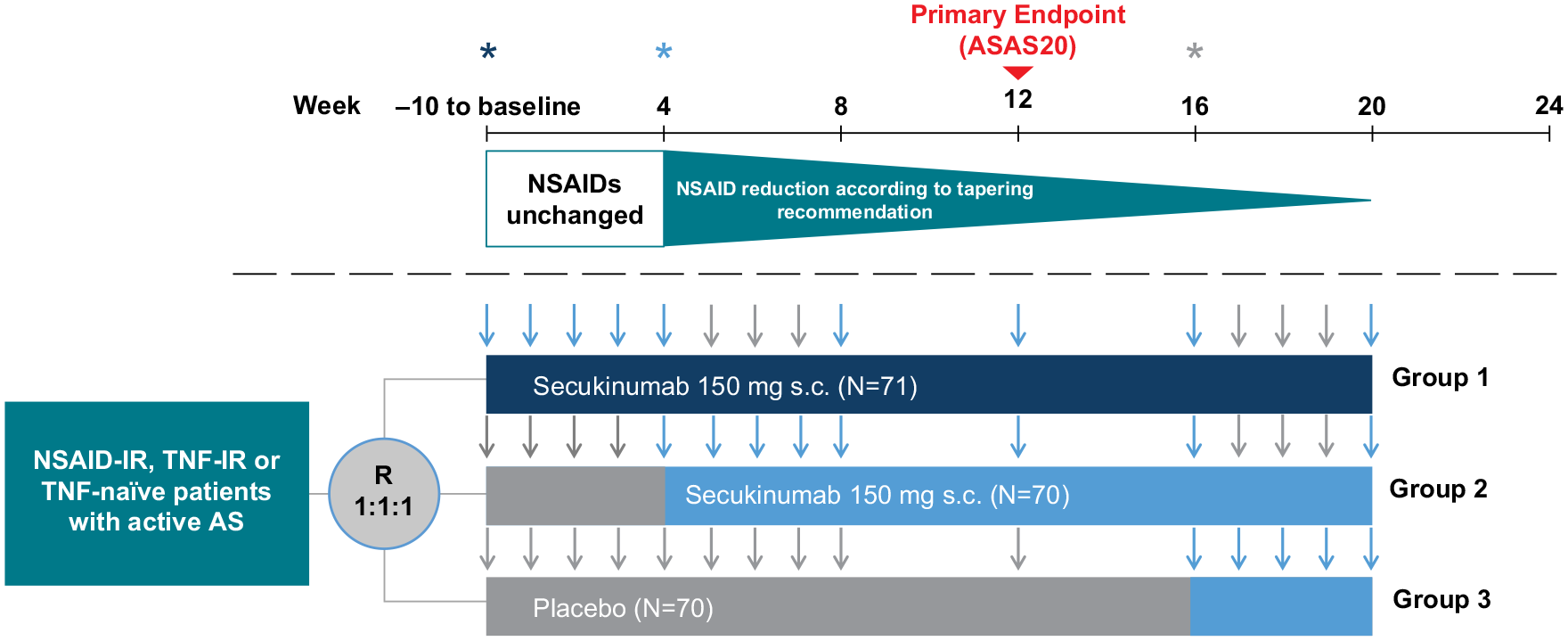
Study design of the ASTRUM study.
Patients were informed that any reduction in the dosage of NSAIDs should be performed gradually and not stopped abruptly upon an improvement of back pain and after discussion with the investigator. Patients were permitted to increase their NSAID dosage on an on-demand basis related to an increase in back pain. All changes in NSAID intake were reported by the patients in a paper-based diary and then, after checking the internal consistency, transferred to the electronic case report form. This was done at all scheduled study visits after week 4. Site visits were at baseline and week 1, and then every 2 weeks from weeks 2 to 8. From week 8 onward, site visits were scheduled every 4 weeks. Patients had options to visit the sites at weeks 3, 5, 7, 17, 18, and 19.
The NSAID-sparing effect of secukinumab was assessed using the ASAS-NSAID score, a standardized tool to evaluate NSAID intake in clinical trials, 34 which ranges from 0 (no intake) to ⩾100 (maximal intake). To calculate the score, the NSAID type, the total daily dose, and the number of days of intake during the period of interest are required. The score was calculated taking both the daily dose of the NSAID used and the percentage of days of intake of the NSAIDs during the period of interest into account. 34
Patients
Patients aged ⩾18 years, with BASDAI and spinal pain (BASDAI question 2) of ⩾4 cm and fulfilling the modified New York criteria for r-axSpA, were enrolled in the study. The patients included had been on at least two different NSAIDs at the highest recommended dose for at least 4 weeks prior to randomization. Patients had to have an inadequate response to NSAIDs within this time period or less if therapy had to be reduced or withdrawn due to intolerance, toxicity, or contraindications.
At screening, all enrolled patients were regularly taking ⩾50% of the highest recommended dose of NSAIDs. After the standardized washout period, patients with prior TNFi therapy reporting regular intake of NSAIDs (50% of the highest recommended dose) at baseline could also be included. At least 2 weeks before randomization, patients had to be on a stable dose of NSAIDs. Patients on a stable dose of methotrexate (⩽25 mg/week) or sulfasalazine (⩽3 g/day) for at least 4 weeks prior to randomization were also allowed to participate in the study.
Patients previously treated with secukinumab or any other biologics directly targeting IL-17 or IL-17 receptor; biological immunomodulating agents; more than two TNFi; high-potency opioid analgesics; and those intolerant to NSAIDs were excluded from this study. Patients with any significant medical condition or another active inflammatory disease were also excluded.
Endpoints
The primary endpoint of this study was the proportion of patients achieving an ASAS20 response at week 12 with secukinumab 150 mg in pooled groups 1 and 2 versus group 3. Higher placebo responses were observed in MEASURE 3, 28 MEASURE 4, 29 and COAST V 35 studies, thereby leading to a potentially smaller difference between active drug and placebo. Therefore, both arms of secukinumab treatment (groups 1 and 2) were pooled to keep sufficient power for the primary and selected secondary analyses. In the initial version of the protocol, groups 1 and 2 had not been pooled.
Secondary outcomes included the mean change from baseline in ASAS-NSAID score for pooled groups 1 and 2 versus group 3 at week 12. The proportion of patients achieving at least 50% reduction in ASAS-NSAID and an ASAS-NSAID score of zero were also assessed.
Exploratory analysis included change from baseline in Ankylosing Spondylitis Disease Activity Score-C-reactive protein (ASDAS-CRP), patient’s global assessment, BASDAI50, and ASAS40. Patient-reported outcomes, using Short-Form Health Survey version 2 (SF-36 v2), and BASDAI were assessed up to week 20 in all three groups. Safety was evaluated in terms of adverse events (AEs) and serious AEs. The reporting of this study conforms to the CONSORT 2010 Statement. 36
Statistical analysis
Sample size was calculated based on the phase III results of the MEASURE 2 study, 27 as these used the same dose and loading regimen as the ASTRUM trial. Using 56.9% ASAS20 responders in the delayed tapering secukinumab group and 27.0% in the placebo group, a sample size of 62 patients per group was required in order to reach 90% power and demonstrate superiority of secukinumab over placebo at a two-sided α = 5%. For the comparison of group 1 at week 16 versus group 3, the same treatment effect was expected, and therefore the same sample size was used. In order to compensate for patient dropout and protocol deviations, the sample size was increased to 68 patients per arm, resulting in a total trial sample size of 204 patients; however, it was only possible to recruit 190 patients for the trial. Therefore, using a final sample size of 190 patients, in a comparison of pooled secukinumab groups versus placebo, the ASTRUM trial had 78% power, assuming a placebo response of 35%. All analyses are presented for the overall population and stratified by TNFi status (naïve or inadequate response) at randomization unless otherwise specified. Regardless of adherence to randomized treatment, the primary endpoint and selected secondary endpoint analyses were based on the intervention effect between secukinumab pooled groups 1 and 2 versus group 3.
Using a linear contrast, the two secukinumab groups were pooled which compared the mean of group 1 and group 2 versus group 3 at week 12 to determine the significance of the secukinumab pooled treatment effect.
Pairwise comparisons of groups 1 and 2 versus group 3, using Fisher’s exact test, were performed up to week 20. In order to determine the significance of the treatment effect of the two secukinumab groups, p values from Fisher’s exact test were displayed up to week 20, prior to placebo re-randomization.
At week 12, logistic regression model with treatment as factor and baseline weight as covariate was used to assess the ASAS20 response for pooled groups 1 and 2 versus group 3 and group 1 versus 3 and separately for group 2 versus 3. Missing data for ASAS20 response and other binary efficacy variables up to week 20 were considered non-responders (non-responder imputation). As there was no sufficient data to calculate sample size and to understand the NSAID-sparing effect of secukinumab during study planning stage, ASAS20 was evaluated as the primary endpoint instead of ASAS-NSAID, which was in line with literature across various r-axSpA studies available at that time where ASAS20 was most accepted endpoint for evaluation.
Mixed models for repeated measures were used to evaluate the differences between treatment groups for the ASAS-NSAID score and HR-QoL relative to baseline. Treatment group and analysis visit were used as factors, and baseline ASAS-NSAID and SF-36 PCS scores and weight were used as continuous covariates.
Results
Patient disposition
Out of 236 patients screened for eligibility, 211 patients (89.4%) were randomly assigned to group 1, group 2, or group 3. A total of 25 patients discontinued the study prior to completion of the screening phase (11.8%), either due to not meeting the eligibility criteria or for other reasons. Out of 211 patients who were randomized, 189 completed 20 weeks of treatment (89.6%). The most frequently used NSAIDs (by more than 10% of patients) were ibuprofen (43.1%), etoricoxib (27.0%), celecoxib (19.0%), and diclofenac (15.6%). The detailed patient disposition through week 20 is presented in Figure 2.

Patient disposition through week 20.
Patient demographics and baseline disease characteristics
The patient demographics and baseline disease characteristics were balanced across groups with a mean age of 45.2 ± 12.3 years, a higher proportion of male patients (57.8%), most (n = 166) being human leukocyte antigen B27 positive (79.0%). The mean time since diagnosis of r-axSpA was 7.4 ± 9.8 years, and the mean BASDAI, ASDAS-CRP score, and ASAS-NSAID score were 6.2 ± 1.4, 3.3 ± 0.73, and 81.7 ± 40.6, respectively (Table 1).
Demographics and baseline disease characteristics in r-axSpA patients.

Group 1: Secukinumab 150 mg initiated from baseline; Group 2: Secukinumab 150 mg initiated from week 4; Group 3: Secukinumab 150 mg initiated from week 16; baseline glucocorticoid intake was balanced in the treatment arms: SEC group 1 15.5%, SEC group 2 12.9%, PBO 13.7%.
ASAS, Assessment of SpondyloArthritis International Society; ASDAS, Ankylosing Spondylitis Disease Activity Score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; BASFI, Bath Ankylosing Spondylitis Functional Index; BASMI, Bath Ankylosing Spondylitis Metrology Index; HLA, human leukocyte antigen; hsCRP, high-sensitivity C-reactive protein; n, number of patients; N, total number of patients randomized; NSAID, nonsteroidal anti-inflammatory drug; PBO, placebo; PGA, patient’s global assessment; r-axSpA, radiographic axial spondyloarthritis; SD, standard deviation; SEC, secukinumab; TNFi, tumor necrosis factor inhibitor.
Efficacy
Even though the primary endpoint of this study was not met, some numerical differences need to be mentioned. For pooled groups 1 and 2 versus group 3, the ASAS20 response rates were 51.1% versus 44.3% (p = 0.35) at week 12 (Figure 3). ASAS40 responses were achieved by a significantly higher proportion of patients in group 1 (p < 0.01) and a numerically higher number of patients in group 2 at week 12 (group 1: 40.8%, group 2: 30.0% versus group 3: 18.6) and week 16 (group 1: 43.7%, group 2: 32.9% versus group 3: 21.4%). Numerically better ASAS40 responses were also observed in groups 1 and 2 patients from week 6 until week 16 compared to group 3 (Figure 3).

Response rates through week 24 in r-axSpA patients. (a) The data shown are the proportion of patients with ASAS20 response. Pooled includes secukinumab 150 mg s.c. group 1 and group 2. Non-responder imputation data through week 24 presented. (b) Descriptive data presented as observed. §p < 0.01 versus placebo. The p value is calculated based on the MMRM, TNFi status, CRP status, and visit as factors, baseline value as continuous covariate, and treatment * visit and baseline value * visit as interaction terms. Arrows indicate first application of secukinumab in the three groups.
In both group 1 and group 2, there was an improvement in the ASAS40 response compared to group 3, despite a reduction in NSAID intake (Figure 4). Furthermore, BASDAI50 was achieved by numerically higher proportion of patients in groups 1 and 2 than in group 3 (group 1: 32.4%, group 2: 28.6% versus group 3: 22.9%). Other secondary outcomes at week 16 are presented in Table 2.

ASAS40 in combination with mean ASAS-NSAID score.
Effect of secukinumab 150 mg s.c. in r-axSpA patients (intention-to-treat population) at week 16.

Observed data (patients) for groups 1, 2, 3, respectively: **67, 66, 62; ***67, 66, 63; ++67, 67, 65; ^20, 19, 20; ^^51, 51, 50.
p Values are from a logistic regression model with treatment, TNFi status (IR/naïve), and CRP status (>/⩽ central laboratory ULN) as factors.
p Values are from MMRM with treatment, TNFi status (IR/naïve), CRP status (>/⩽ central laboratory ULN), and visit as factors and baseline value as a continuous covariate. Missing values were imputed as non-response.
p < 0.001, §p < 0.01, and ‡p < 0.05 versus group 3; ‡‡p < 0.05 versus group 2.
ASAS, Assessment of SpondyloArthritis International Society; ASDAS, Ankylosing Spondylitis Disease Activity score; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; CRP, C-reactive protein; IR, inadequate responder; NSAID, nonsteroidal anti-inflammatory drug; N, total number of patients in each treatment group; PR, partial remission; r-axSpA, radiographic axial spondyloarthritis; SD, standard deviation; sec, secukinumab; TNFi, tumor necrosis factor inhibitor.
A numerically higher proportion of TNFi-naïve patients in group 1 and a significantly higher number of patients in group 2 achieved an ASAS20 response compared to group 3 (group 1: 54.9%, group 2: 58.8% versus group 3: 40.0%). Furthermore, a significantly higher proportion of TNFi-IR patients in group 1 achieved an ASAS20 response (group 1: 60.0%, group 2: 26.3% versus group 3: 45.0%). In both groups, 1 and 2, a significantly higher proportion of TNFi-naïve patients achieved an ASAS40 response compared to group 3 in Table 2. No significant improvement of ASAS40 was noted in TNFi-IR patients.
The mean change in BASDAI score in comparison to baseline was significant only in group 1. In both groups 1 and 2, the mean change in ASDAS-CRP was significantly higher compared to group 3 [group 1: −1.2 (0.9), p < 0.01, group 2: −1.0 (0.9), p < 0.05 versus group 3: −0.7 (0.8)] (Table 2).
A decrease in the ASAS-NSAID scores after 4 weeks was observed in all three treatment groups, which continued till week 16. At week 12, a marked reduction in the ASAS-NSAID score was noted in all treatment groups (group 1: −44.9 ± 47.3; group 2: −40.3 ± 71.5; group 3: −31.5 ± 36.5) but was more pronounced in groups 1 and 2.
A significantly higher proportion of patients in group 1 and a numerically higher proportion of patients in group 2 showed a reduction of the NSAID score at week 16 of ⩾50% compared to group 3 (group 1: 64.8%, group 2: 58.6%, and group 3: 42.9%; p < 0.05) (Table 2 and Figure 5). In both groups 1 and 2, the number of patients with a reduction of the NSAID score < 10 was significantly higher as compared to group 3. At week 16, a significantly higher number of patients in groups 1 and 2 versus group 3 completely stopped NSAID intake (group 1: 32.4%, p < 0.05; group 2: 38.6%, p < 0.01 versus group 3: 17.1%) (Table 2 and Figure 5).

Nonsteroidal anti-inflammatory drug-sparing effects. (a) ASAS-NSAID score, mean change from baseline to week 24 in the secukinumab and placebo group. Statistical analysis is exploratory, as the primary endpoint was not met. Observed data is presented using descriptive statistics. †p < 0.001; §p < 0.01; and ‡p < 0.05 versus placebo; p value are from MMRM model with treatment and week after baseline as factors, weight and baseline ASAS-NSAID score as continuous covariates, treatment * week after baseline and baseline ASAS-NSAID * week after baseline as interaction terms in the model terms. (b) Proportion of patients with ASAS-NSAID score reduction of ⩾50% through week 24. Observed data is presented using descriptive statistics through week 24. (c) Proportion of patients reaching ASAS-NSAID score = 0 through week 24. Observed data is presented using descriptive statistics through week 24. Statistical analysis is exploratory, as the primary endpoint was not met; p values are from a logistic regression model with treatment, TNF inhibitor status (inadequate responder/naïve), and CRP status (>central laboratory ULN/⩽laboratory ULN) as factors. †p < 0.001, §p < 0.01, and ‡p < 0.05 versus group 3.
In groups 1, 2, and 3, the mean ± SD change in ASAS-NSAID score was −51.5 ± 46.2, −42.5 ± 68.8, and −33.7 ± 38.8, respectively (Figure 5). A significantly higher proportion of patients in group 1 showed a reduction in the ASAS-NSAID score compared to group 2 (p < 0.05). The reduction in NSAID intake continued in all groups after week 16. The NSAID dose reduction in group 3 until week 24 was comparable to groups 1 and 2. Between these two groups there was no major difference in mean NSAID reduction (Table 2 and Figure 5). The ASAS-NSAID score at week 20 was comparable in all groups, as shown in Figure 5.
Patient-reported outcomes showed noticeable improvements at week 12, especially in group 1 and group 2, as assessed by changes in SF-36 PCS scores (41.7 ± 8.33 versus 40.6 ± 6.64 in groups 1 and 2 versus group 3, respectively).
Safety
In the overall population, mean duration of exposure to study treatment (secukinumab 150 mg and placebo) was 110.2 days (63.64 patient-years in total): 110.3 days (21.43 patient-years), 111.3 days (21.33 patient-years), and 108.9 days (20.87 patient-years) for group 1, group 2, and group 3, respectively. Safety results up to week 16 are presented in Table 3.
Safety profile up to week 16.

AE, adverse event; N, total number of patients randomized; n, number of patients with AE; SD, standard deviation.
Up to week 16, incidence of any AEs and serious AEs was 78.2% and 3.8% in the overall population while AEs leading to treatment interruption or discontinuation 2.8%.
Overall, 7.1% of the study population presented with a history of inflammatory bowel disease (IBD) at baseline. Patients with active IBD were excluded from the study. A total of 5.7% of patients reported IBD (broadly defined) from baseline to week 16; these included three cases of diarrhea, two cases of colitis, one case of Crohn’s disease, and one case of diarrhea hemorrhagic, the latter two both in the SEC-DT group. In both cases, the outcome was reported as resolved/recovered. While after week 16 IBD (diarrhea) was reported in 2.4% of patients. There was no obvious difference between the three study groups, with an overall low incidence rate of IBD.
In all three groups, uveitis, Crohn’s disease, and depression were reported as the selected AEs of interest up to week 16. No deaths were reported during the study.
Discussion
Therapy with NSAIDs is recommended as first-line treatment for patients with axSpA, 37 with the vast majority of them receiving these drugs as the earliest pharmacological intervention to improve their back pain. 6 However, there is still debate on the long-term benefit of NSAIDs due to (i) their limited therapeutic effect, (ii) frequent intolerance reported by patients, (iii) the substantial risk of associated AE, and (iv) bad adherence.38,39 Furthermore, in almost one-third of patients with rheumatic and musculoskeletal diseases, NSAIDs may be inappropriately prescribed. 9 Accordingly, in the recent EULAR recommendations on early arthritis management, NSAIDs are recommended as effective symptomatic therapies only after evaluating gastrointestinal, renal, and cardiovascular risks and at the minimal effective dose for the shortest possible time. 40 However, this differs in axSpA for several reasons, including the younger age of patients in early disease in comparison to other rheumatic and musculoskeletal diseases, such as rheumatoid arthritis. 37
In view of the potential risks associated with long-term use of NSAIDs, clinicians favor discontinuation or tapering of NSAIDs once a sufficient clinical response (i.e. remission of low disease activity) on treatment with bDMARDs has been achieved.17,38 However, only a few trials have assessed the NSAID-sparing effects of bDMARDs in r-axSpA to date.2,17,23
To our knowledge, this is the first study to systematically determine the short-term NSAID-sparing effect in patients treated with secukinumab or placebo following an initial run-in phase of NSAID therapy administered in a stable dosage. However, a long-term sustained NSAID-sparing effect of secukinumab maintenance dosages of 150 and 300 mg in patients with r-axSpA has been recently reported in a post hoc analysis of data pooled from several MEASURE studies. 26
The efficacy data reported in this study (ASTRUM group 2) are largely comparable to those from the secukinumab 150 mg arm of the MEASURE 2 study. 27 However, the placebo scores of this study were lower than those in the ASTRUM study. Compared to the secukinumab 150 mg arm of MEASURE 3 and 4, the ASAS20 and ASAS40 response rates were similar to those in ASTRUM.28,29 The relatively high placebo response rate observed in this study could be due to a number of reasons, some of which have been discussed in the available literature.30–32 There are many factors involved which might play a role and influence the treatment outcome: age, sex, disease duration, a high degree of disease activity and pain, impaired function and mobility, personality traits and ethnicity, compliance, additional medication, physiotherapy, and physical activity.30–32 Many of these factors could not be or were not controlled for in the ASTRUM study, for example, in this study, the patients were older than those in the MEASURE 2–4 studies. This might have impacted treatment effects, as younger patients show better treatment responses27–29,41; an effect which is also reported for TNFi. 42 In addition, more females were included in the ASTRUM study than in the MEASURE studies; however, this is in line with German national epidemiological data. 43 Furthermore, patients in the ASTRUM study had prior knowledge about the approved status of secukinumab in r-axSpA, and all patients included in the study were aware that treatment with verum would commence from week 16 at the latest. Thus, a positive effect on treatment response rates is plausible.
Importantly, there was evidence for a greater decrease in NSAID intake, as documented by the marked reduction in ASAS-NSAID scores. Thus, the NSAID intake was lowered between weeks 4 and 16, and this was more pronounced in groups 1 and 2 compared to group 3. At week 20, there was no major difference between tapering group 1 and group 2, with comparable NSAID scores across all three groups at week 20. The decrease in NSAID intake seemed to be independent of the dosing regimen and the time of administration of secukinumab. This suggests that tapering of NSAIDs might possibly be initiated at any time during secukinumab therapy, depending on physician’s assessment (disease activity, comorbidities) and patient’s preferences.
The results of our study are largely consistent with data from the SPARSE and TOPAS trials, in which the intake of NSAIDs was also reduced in patients treated with other bDMARDs.17,23 Since an improved efficacy was seen even in the former placebo group between weeks 16 and 20, upon active treatment with secukinumab, tapering of NSAIDs may be attempted before initiation of secukinumab treatment. However, considering the high placebo response in this study, other explanations cannot be ruled out.
In the absence of scientific evidence regarding the optimal timing of NSAID tapering, the results of ASTRUM will hopefully help rheumatologists and practitioners to taper and discontinue NSAIDs in time. Importantly35.8% of patients in groups 1 and 2 did not require more NSAIDs after treatment with secukinumab for 12 weeks. At week 16, more than 30% of patients in all three study groups were no longer taking NSAIDs at all.
Even though NSAID intake could be successfully reduced, the efficacy of secukinumab therapy remained high. Therefore, we hope that the findings of this study will guide rheumatologists to adjust concomitant NSAID treatment in patients with r-axSpA. This may also help the optimization of treat-to-target strategies 6 based on therapy with secukinumab.
However, our study has certain limitations. Firstly, due to the limited sample size used in our study, as well as the inclusion of patients with TNF intolerance in the TNF-IR group, we did not separately examine primary and secondary non-responders to anti-TNFs. In addition, no exact recommendation could be given as to best time point of the tapering of NSAIDs. Finally, in this study, AEs could not be unequivocally attributed to study the drug or to concomitant NSAID treatment.
Conclusion
Even though the primary endpoint of our study was not met, it can be concluded that secukinumab treatment in patients with r-axSpA demonstrated short-term NSAID-sparing effects along with improvements in predefined clinical outcomes. Furthermore, this study highlights the benefits of an early initiation of secukinumab which may led to discontinuation of NSAIDs in more than 30% of patients without affecting secukinumab response rates. Overall, secukinumab was well tolerated and no unexpected new safety signals occurred.
Footnotes
Appendix
Acknowledgements
The authors thank all patients who participated in the study, the clinical investigators, and study personnel for their willingness to participate in the study. The authors also thank Andreas Haehle, Novartis Pharma GmbH, Germany, for medical guidance and editorial support. Manuscript writing support was provided by Dhanya Mukundan and Shravanti Mukherjee (Novartis Healthcare Pvt Ltd, Hyderabad, India) and Kate Killick (Novartis Ireland Ltd). The data from this study were partially presented at EULAR 2021.