
Abstract
In spite of a major public health burden with increasing prevalence, current osteoarthritis (OA) management is largely palliative with an unmet need for effective treatment. Both industry and academic researchers have invested a vast amount of time and financial expense to discover the first diseasing-modifying osteoarthritis drugs (DMOADs), with no regulatory success so far. In this narrative review, we discuss repurposed drugs as well as investigational agents which have progressed into phase II and III clinical trials based on three principal endotypes: bone-driven, synovitis-driven and cartilage-driven. Then, we will briefly describe the recent failures and lessons learned, promising findings from predefined post hoc analyses and insights gained, novel methodologies to enhance future success and steps underway to overcome regulatory hurdles.
Introduction
Impact and burden of osteoarthritis
Osteoarthritis (OA) is the most prevalent age-related articular disease and commonly manifests in the ageing population with a spectrum of signs and symptoms such as chronic articular pain, brief morning stiffness, limitation of functional activities, joint line tenderness, bony enlargement, joint deformity, coarse crepitus and muscle wasting. 1 Its global prevalence is estimated at 22.9% in persons over 40 years of age in 2020 (correspondingly 654.1 million individuals). 2 Increased prevalence in the next decades is expected due to the increasing age of the population and the obesity epidemic. Furthermore, the socioeconomic costs of OA are considerable 3 as 1–2.5% of the gross national product (GNP) has to be utilized as the direct and indirect costs for OA management in most Western countries, 4 constituting a major public health challenge for coming decades.
Drug development and clinical unmet needs
Current OA management focuses on symptomatic improvement only 5 and is largely palliative, although OA disease course is usually progressive over many years. 6 In addition, the existing pharmacological or non-pharmacological treatments have shown only modest efficacy at best 7 and adverse effects in the gastrointestinal (GI), cardiac or renal systems prohibit the long-term use of commonly used analgesics as OA patients are often elderly with comorbid diseases. 1 Hence, there exists an immense unmet need in the current therapies, with more than half of the patients with moderate and severe OA reporting unsatisfactory pain relief. 8
Despite such an enormous impact and disease burden on OA patients, no effective disease-modifying osteoarthritis drugs (DMOADs) have been approved by regulatory bodies. 9 A DMOAD is a pharmacological agent that will delay or reverse the progression of the structural damage of the joint, thereby leading to clinical translation of improvement in symptoms, manifested either by pain reduction or benefits in physical function. 9 Therefore, both structural and symptomatic benefits are needed for an agent to be considered a DMOAD. 10 It is crucial to discover innovative, effective DMOAD therapies to mitigate the disease burden.
Phenotypes/endotypes in OA
OA is a heterogeneous and multifaceted disease that manifests first as a molecular derangement (abnormal metabolism in the joint tissue) leading to anatomic and/or physiologic malfunction such as osteophyte formation, cartilage damage, synovitis and loss of normal joint function that can culminate in illness and whole joint organ failure. 11 Therefore, classification of the OA patients into subgroups that possess distinct characteristics or pathogenic pathways will enable clinical trials to identify appropriate patients who may have benefits from a particular investigational agent. Broadly, OA disease can involve different ‘clinical phenotypes’ or ‘molecular endotypes’. The term ‘clinical phenotype’ should represent the clinical manifestations of a disease and is based on observable traits such as aetiologic factors and risk factors, whereas ‘molecular endotype’ should pertain to the molecular drivers of disease pathogenesis via cellular, molecular and biomechanical signalling pathways and is irrespective of its clinical manifestations. 12
As an important qualification, OA patients may have overlapping clinical phenotypes, and a given clinical OA phenotype may possess a variety of molecular endotypes at different stages of pathogenesis pathways, complicating the task of identifying the distinct OA subgroups.9,13 However, generally, a consensus seems to exist regarding the presence of three endotypes (Figure 1): (1) bone-driven endotype, (2) synovitis-driven endotype and (3) cartilage-driven endotype.9,14
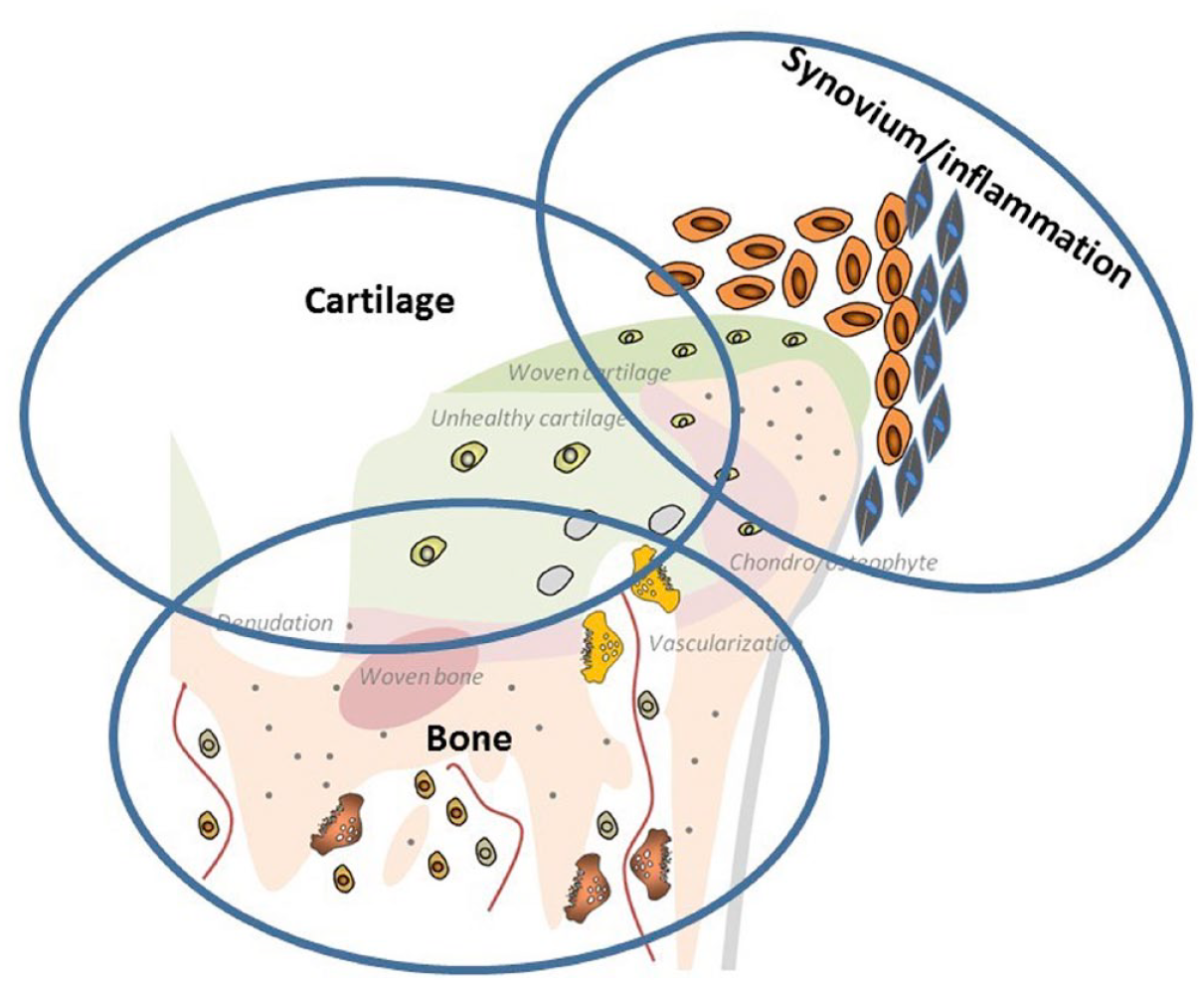
The three endotypes of OA: (1) bone-driven endotype, (2) synovitis-driven endotype and (3) cartilage-driven endotype.
Bone-driven endotype
An uncoupled remodelling process between bone formation and resorption in the subchondral bone results in microstructural changes, depending on the spontaneous stimulation (in early-stage OA) or inhibition (in late-stage OA) of osteoclastic bone resorption. 15 Furthermore, bone pain can be induced by an acidic microenvironment created via H+ ions which bone-resorbing osteoclasts generated as shown in animal models of bone metastasis. 16 Osteoclasts can produce Netrin-1, leading to sensory innervation and pain via its receptor DCC (deleted in colorectal cancer). 17 These findings indicate the potential of bone-protective agents in DMOAD research.
Synovitis-driven endotype
In OA patients, there is biochemical and histological evidence 18 of infiltration of mononuclear cells in the synovium, 19 proliferation of synovial cells as well as inflammatory cytokines such as tumour necrosis factor (TNF)-α, interleukin (IL)-1β and IL-6 20 supported by the findings with imaging modalities. 21 In addition, an association of synovitis with symptoms and radiological progression was reported, 22 suggesting the potential role of anti-inflammatory agents in an OA subgroup with predominant inflammatory changes.
Cartilage-driven endotype
The hallmark of OA pathogenesis seems to be a failure to maintain cartilage homeostasis resulting in an imbalance between synthesis and degradation of extracellular matrix components. This phenomenon can lead to cartilage softening, fibrillation and fissuring of the superficial cartilage layers, and diminished cartilage thickness. 23
Our review will focus on the DMOAD candidates currently undergoing or having completed the active phase II and III clinical trials since 2017 by conducting electronic and manual searches on the https://clinicaltrials.gov/ (Tables 1 and 2). Although the assignment of a specific drug on account of its predominant activity was made only to one specific endotype, some drugs may have broader endotype effects, and where present, these are duly described.
Summary of repurposed DMOAD clinical trials which are active or completed since 2017 (phases II and III).

DMARD, disease-modifying antirheumatic drug; DMOAD, disease-modifying osteoarthritis drug; IL, interleukin; IV, intravenous; OA, osteoarthritis; PTH, parathyroid hormone; S/C, subcutaneous; TNF. tumour necrosis factor.
Summary of investigational DMOAD clinical trials which are active or completed since 2017 (phases II and III).

ADAMTS, a disintegrin and metalloproteinases with thrombospondin motifs; ANGPTL3, angiopoietin-like 3; DMOAD, disease-modifying osteoarthritis drug; FGF, fibroblast growth factor; IA, intra-articular; IL, interleukin; OA, osteoarthritis.
Recent phase II and III clinical trials
These agents will be broadly divided into two major groups: (1) repurposed drugs and (2) investigational drugs. The individual drugs included are illustrated in Figure 2.

Repurposed or invesigational drugs related to the three main OA endotypes (phase II and III RCTs).
Repurposed drugs already in phase II and III trials, either active or completed since 2017
The costs of bringing new drugs to the market increased ninefold from 1979 (US$92 million) to 2010 (US$883.6 million), 24 and were reported at US$1335.9 million (in 2018 US dollars). 25 Due to high attrition rates, escalating costs and regulatory hurdles, drug repurposing strategy is often used for rediscovering new uses for existing approved therapeutic agents outside the scope of the original indication. 26 This approach offers a better risk-versus-reward trade-off over the de novo drug discovery as the safety and pharmacokinetic profiles have already been examined in preclinical models and humans, and the time frame for drug development is much reduced. 27 This leads to substantial savings in preclinical and phase I and II costs, 28 and reduces the development cost to US$300 million on average. 29 Repositioning success stories were evidenced in history (e.g. sildenafil in erectile dysfunction 30 ) and recent pharmacological treatment for COVID-19 disease. 31 With the proposed biochemical pathway scientifically validated, several existing drugs have been examined for the repositioning idea, some of which are in advanced stages of OA clinical trials (Table 3).
Published results of phase II/III clinical trials related to the repurposed DMOAD agents since 2017 by OA endotypes.

denotes P ≤ 0.05, active vs control. AIMS2-SF, Arthritis Impact Measurement Scale 2–Short Form; AUSCAN, Australian Canadian Hand Osteoarthritis Index; BML, bone marrow lesion; CI, confidence interval; DMOAD, disease-modifying osteoarthritis drug; FIHOA, Functional Index for Hand Osteoarthritis; GUSS, Ghent University Scoring System; HCQ, hydroxychloroquine; IM, intramuscular; IV, intravenous; KOOS, Knee injury and Osteoarthritis Outcome Score; MRI, magnetic resonance imaging; MTX, methotrexate; NA, non-available; NRS, Numerical Rating Scale; OA, osteoarthritis; SC, subcutaneous; SD, standard deviation; VAS, Visual Analogue Scale; WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index.
Bone-driven endotype
Antiresorptive agents, which are administered as the standard treatment in osteoporosis, 44 may be promising therapies as subchondral remodelling on which these agents act is involved in OA pathogenesis, including magnetic resonance imaging (MRI)–detected bone marrow lesions (BMLs). 45 BMLs are observed as an altered signal pattern related to increased vascularization, bone marrow necrosis, fibrosis and less mineralized bone on MRI 46 and as an abundance of matrix metalloproteinases (MMPs), TNF-α and substance P on histochemical analysis. 47 BMLs have an established association with OA pathogenesis and symptoms.48,49 In addition, compared with normal joints, the elastic modulus of the trabecular subchondral bone was reduced by 60% in early OA, 50 perhaps due to incomplete mineralization sequence because of the high rate of bone remodelling. 51 Therefore, antiresorptive agents seem to be a promising therapy due to their implications in pathogenesis and clinical manifestations in OA. Recently, teriparatide, clodronate, zoledronic acid, denosumab and vitamin D have been investigated as the potential agents targeted at the bone-driven OA endotypes.
Teriparatide
Teriparatide is a recombinant human parathyroid hormone (PTH) and acts on bone-forming osteoblasts, resulting in an anabolic effect within active remodelling sites (remodelling-based osteogenesis), and on surfaces of previously inactive bone (modelling-based osteogenesis). 52 Following completion of the Fracture Prevention Trial (FPT), 53 it was granted approval by the US Food and Drug Administration (FDA) in 2002 and later by the European Medicines Agency (EMA) in 2003. 54 It showed an increase in proteoglycan content, reducing cartilage damage in OA animal models 55 as well as pain reduction in temporomandibular joint OA changes in ageing mice. 56 Intermittent administration of teriparatide improved pain by reducing prostaglandin E2 and sensory innervation of subchondral bone. 57 A phase II clinical trial in knee OA is currently ongoing (NCT03072147).
In the context of long-term OA treatment, one of the potential safety issues related with teriparatide is osteosarcoma due to toxicology findings of osteosarcoma in rats. 58 However, administering non-human primates (cynomolgus monkeys) with the drug as eight times large as the dose used in the human dose did not lead to osteosarcoma development. 59 In addition, recent analysis of Forteo Patient Registry based on the estimated 242,782 person-years of observation 60 as well as a 15-year US Postmarketing Surveillance Study 61 revealed no incident cases of osteosarcoma among teriparatide users after 8 years of follow-up. In November 2020, FDA approved labelling changes such as removing the 2-year limitation and the boxed warning about the osteosarcoma risk. 62
Clodronate
Clodronate is a first-generation non-nitrogenous bisphosphonate and possesses anti-inflammatory and analgesic effects 63 in addition to osteoclast inhibition and apoptosis. 64 It reduced the generation of cytokines (IL-1, TNF-α) and metalloproteases (MMP 2/3/9) in the synovium and cartilage in animal models. 65 In a small study including 74 patients with knee OA, a reduction of pain and BMLs was reported after providing a higher dose of intramuscular clodronate than used for osteoporosis (200 mg daily for 15 days and then once weekly for the next 11.5 months), compared with a shorter maintenance regimen (2.5 months). 32 In non-overweight individuals in whom weight-bearing damage is not a driving cause of OA progression, a bone-related abnormality and remodelling may play a crucial role. 66 Non-overweight female patients (body mass index, BMI < 25 kg/m2) with early radiographic knee OA [baseline Kellgren-Lawrence (KL) grade <2] revealed a 51% reduction of 2-year radiographic progression after bisphosphonate exposure by utilizing a propensity-matched retrospective cohort analysis of the Osteoarthritis Initiative (OAI; n = 346) with no significant effects in patients with advanced OA disease. 67
Zoledronic acid
Zoledronic acid is a third-generation nitrogen-containing bisphosphonate, and inhibits farnesyl pyrophosphate synthase, which is critical for osteoclast function. 68 In in vitro studies, its inhibition is 3-, 7-, 17- and 67-fold more potent than risedronate, ibandronate, alendronate and pamidronate, respectively.69,70 It also showed the highest binding affinity for the hydroxyapatite preferentially at sites of high bone turnover. 71 It was first approved in 2002 for adjunctive treatment for multiple myeloma and bone metastases to reduce skeletal-related events 72 and later in 2007 for postmenopausal osteoporosis, 73 based on the findings of the landmark Health Otcomes and Reduced Incidence with Zoledronic Acid Once Yearly (HORIZON) Pivotal Fracture Trial. 74
In a recent 2-year Zoledronic Acid for Osteoarthritis Knee Pain (ZAP2) study targeted at the bone-driven endotypes, knee OA patients with significant knee pain and MRI-detected BMLs were recruited and provided with twice yearly administration of 5 mg of zoledronic acid (n = 223). Despite methodological strengths such as adequate power to detect disease-modifying effects on BMLs, utilization of the more sensitive MRI-detected cartilage volume as the primary outcome, and the 2-year length of the follow-up, the study failed to reveal no significant improvement in knee pain, BML size and cartilage volume loss. 33 A greater symptomatic improvement was detected in early knee OA patients [i.e. without radiographic joint space narrowing (JSN)] on the exploratory subgroup analysis. 33 There is neither structure-modifying benefit nor reduction of knee replacement (KR; 9% in the zoledronic acid group versus 2% in the placebo group). Currently, one more clinical trial is examining the effects of Zoledronic Acid in hip OA (NCT04303026).
Denosumab
Denosumab is a fully human monoclonal immunoglobulin G2 (IgG2) antibody that binds and inhibits the receptor activator of nuclear factor-κB ligand (RANKL), which selectively inhibits osteoclastogenesis with a profound decrease in the rate of bone remodelling. 75 Its use for postmenopausal osteoporosis was approved by the FDA in the United States and by the European Medicines Agency in Europe since 2010, 76 based on the results of the pivotal Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) trial. 77 In terms of bone density and remodelling, denosumab is more efficacious than the alendronate, risedronate and ibandronate. 78 Denosumab might lead to a dramatic increase in bone mass, perhaps through anabolic action on bone remodelling. 79 However, there is no clue regarding the effect of this phenomenon on the quality of subchondral bone in early OA patients. 50 There is one clinical trial evaluating the benefits of denosumab in hand OA (NCT02771860).
Vitamin D
Vitamin D is a fat-soluble vitamin that is essential for calcium homeostasis and bone metabolism, such as subchondral remodelling. Both early-stage increased remodelling and bone loss, and the late-stage slow remodelling and subchondral sclerosis are implicated in different stages of the OA pathogenetic process. 80 It promotes proteoglycan production in chondrocytes and reduces the release of inflammatory cytokine via stimulation of 5′-adenosine monophosphate-activated protein kinase (AMPK)/mTOR signalling. 81 In a Vitamin D and Omega-3 Trial (VITAL) in patients with chronic knee pain, vitamin D supplementation showed no improvement in both pain and function at 4-year follow-up. 35 Vitamin D supplementation provided for symptomatic knee OA caused no structural benefits in the MRI outcomes such as synovial volume and subchondral BML volume at 2-year follow-up (n = 50). 34 In a recent meta-analysis, there is no structure-modifying effect of vitamin D in knee OA. 82 A small phase IV clinical trial is ongoing (NCT04739592).
Synovitis-driven endotype
A variety of conventional and biological disease-modifying antirheumatic drugs (DMARDs), which are being widely utilized in the treatment of inflammatory arthritides such as RA83,84 and psoriatic arthritis, 85 were recently examined in patients with inflammation-driven OA endotypes due to their impressive anti-inflammatory action. In a meta-analysis published in 2018, DMARDs failed to show clinically significant symptomatic improvement in either hand (both erosive and non-erosive) or knee OA. 86 Recently, methotrexate (MTX), hydroxychloroquine (HCQ), etanercept and tocilizumab were examined for repurposing agents in OA.
Methotrexate (MTX)
MTX was studied initially as a chemotherapeutic agent for childhood leukaemia in 1948 87 and then in rheumatoid arthritis (RA) around 1970, 88 leading to FDA approval as a conventional DMARD in 1988. 89 RA is a prototype of chronic inflammatory autoimmune disease with florid inflammation of the synovial joints involving proinflammatory cytokines such as IL-1 and TNF-α. 90 MTX is a folate analogue that binds dihydrofolate reductase to interfere with DNA synthesis in actively dividing cells and induces inhibition of IL, TNF-α and so on. 91 Currently, MTX is the anchor drug for RA treatment.
As the synovitis-driven OA endotype has increased levels of proinflammatory cytokines, modulating the inflammatory response by using MTX may be beneficial theoretically. In the phase III PROMOTE trial published as a 2019 Osteoarthritis Research Society International (OARSI) conference abstract (n = 134), oral MTX showed significant improvement in knee pain and function at 6-month follow-up but not at 9 and 12 months with no change in synovial volume on contrast-enhanced MRI at 6 months. 36 A recent study on 64 participants with symptomatic erosive hand OA revealed no significant difference over the pain VAS (Visual Analogue Scale) score and functional outcomes on Functional Index for Hand Osteoarthritis [FIHOA] score at both 3- and 12-month follow-ups. 37 There was also no significant difference in structural progression evaluated by plain radiographs and MRI BMLs, erosions and synovitis. Currently, a phase III knee study is active for symptomatic OA patients with effusion-synovitis grade of ⩾2 (NCT03815448).
Hydroxychloroquine (HCQ)
HCQ belongs to the group of antimalarial agents, was synthesized in 1946 and is currently used in various rheumatic and skin diseases since its first approval in 1955 by FDA.92,93 It possesses anti-inflammatory actions such as decreasing cytokine production from T cells and monocytes, especially IL-1 and IL-6. 94 In the first randomized controlled trial (RCT) conducted in hand OA and published in 2018, administration of oral 400 mg HCQ once a day for 24 weeks is not superior to placebo in improving symptomatic hand OA at weeks 6, 12 and 24 (n = 196). 38 Similarly, another bigger HERO study revealed no significant treatment differences at 3, 6 or 12 months in hand OA (n = 248). 39 Recently, in the OA-TREAT clinical trial conducted in erosive hand OA for 52 weeks (n = 153), HCQ failed to show any difference in symptomatic and radiographic outcomes, confirming the findings of the previous two studies. 40
Etanercept
Etanercept was the first TNF inhibitor approved to treat RA in the United States in 1998 and Europe in 2000. 95 In a 1-year, Etanercept in patients with inflammatory hand osteoarthritis (EHOA) clinical trial in participants with ⩾4 interphalangeal joints (IPJs) with osteoarthritic nodes, ⩾1 IPJ with soft tissue swelling or erythema, ⩾1 IPJ with positive power Doppler activity on ultrasound and ⩾1 IPJ with radiographic pre-erosive or erosive disease (n = 90), etanercept (24 weeks 50 mg/week, thereafter 25 mg/week) did not improve pain and function at 24 weeks or 1 year. However, on subgroup analyses of participants with active inflammation, such as the presence of soft tissue swelling or power Doppler signals, etanercept revealed an improvement in radiographic scores 41 measured by the validated Ghent University Scoring System. 96 In addition, etanercept reduced serum MMP3-levels but not other soluble biomarkers of inflammation, cartilage and bone damage. 97
Tocilizumab
Tocilizumab is a genetically engineered humanized neutralizing antibody inhibiting the binding of IL-6 to its receptors 98 and blocking both classic (anti-inflammatory) and trans-signalling (proinflammatory) pathways. 98 Its use in RA was approved in Europe in 2009 and in the United States in 2010. 96 However, in a recent 12-week phase III clinical trial (n = 83), two infusions 4 weeks apart (weeks 0 and 4) of tocilizumab (8 mg/kg intravenous) revealed no significant benefits in pain and function in refractory hand OA with at least three painful joints, compared with the placebo. 42 This might suggest that removing IL-6 signalling alone in the short term is not sufficient for pain reduction in human OA.
Agents with other mechanisms
Biguanides (metformin)
Metformin is a member of the biguanides and the first-line therapy for type 2 diabetes mellitus (DM) patients with obesity. While it was clinically available in the United Kingdom in 1958 and in Canada in 1972, the FDA approved metformin in 1994 for type 2 DM. 100 In murine arthritis models, administration with metformin showed a reduction of arthritis scores and bone destruction as well as an anti-inflammatory effect by lowering serum levels of proinflammatory cytokines mediated via indirect activation of AMPK pathways.101,102 Treatment with metformin increased antinociceptive activity, and anti-inflammatory and chondroprotective effects in OA mice with monosodium iodoacetate (MIA) model 103 as well as with collagenase-induced osteoarthritis (CIOA) model.104,105
A combination of metformin with COX-2 inhibitors (n = 968) reduced the risk of joint replacement surgery (12.81% versus 16.22%) over 10 years compared with COX-2 inhibitors alone (n = 1936) in type 2 DM patients with OA in a nationwide, retrospective, matched-cohort study. 106 However, data on the OA severity, disease duration and other assessment scores are unavailable, limiting the validity of study findings. In OAI participants with radiographic knee OA and obesity (BMI ⩾ 30 kg/m2), metformin users (n = 56) showed slower MRI-detected cartilage volume loss (0.71% versus 1.57% per annum) in the medial compartment of the joint over 4 years compared with non-user (n = 762) despite no significant symptomatic improvement. 107 Currently, the first two RCTs are ongoing (NCT04767841, NCT05034029).
Liraglutide
Liraglutide is a glucagon-like peptide-1 (GLP-1) that regulates glucose and energy homeostasis via GLP-1R binding. Liraglutide was approved by the FDA in 2014, and EMA in 2015, for chronic weight management in overweight people with a BMI ⩾ 27 kg/m2. 108 It attenuated cartilage degeneration in an OA model of knee joints in vivo by exerting antiapoptotic and anti-inflammatory effects on chondrocytes.109,110 Administration of Liraglutide 3 mg/day for 52 weeks in participants with KOA and overweight/obesity (BMI ⩾ 27 kg/m2) revealed no difference in pain between the liraglutide and placebo group despite a significant loss in body weight (n = 156). 43 More GI adverse events were reported in the liraglutide group (50.2% versus 39.2%).
Investigational DMOAD agents already in phase II and III trials, either active or completed since 2017
In the United States, federal law required that a drug must have proven safety as well as efficacy before marketing for the prescription. 111 If a drug appears promising in preclinical studies in a laboratory, a drug sponsor or sponsor-investigator can submit an investigational new drug (IND) application for collecting the data required to establish that the product will not expose humans to unreasonable risks when used in limited, early-stage clinical studies. 112 Therefore, an investigational drug is an experimental drug that is being examined in clinical trials to detect whether it is safe in humans and effective for a particular disease.
Very recently, we have extensively reviewed these investigational agents, and we will briefly discuss and update, if new findings are published in the interim, these drugs in this article (Table 4). Therefore, the readers who are keen on a more detailed coverage are suggested to read the recent reviews.9,13
Published results of phase II/III clinical trials related to the investigational DMOAD agents since 2017 by OA endotypes.

, **, and *** denote P ≤ 0.05, P ≤ 0.01, and P ≤ 0.001, respectively, active vs control. BD, bis die; BML, bone marrow lesion; cMFTC, central medial femorotibial compartment; DMOAD, disease-modifying osteoarthritis drug; IA, intra-articular; IKDC, international knee documentation committee; JSW, joint space width; LFTC, lateral femorotibial compartment; MFTC, medial femorotibial compartment; MRI, magnetic resonance imaging; NA, non-available; NRS, Numerical Rating Scale; OA, osteoarthritis; OD, omne in die; SD, standard deviation; VAS, Visual Analogue Scale; WOMAC, Western Ontario and McMaster Universities Osteoarthritis Index. WORMS, whole organ magnetic resonance imaging score.
Bone-driven endotype
Cathepsin K inhibitor
Cathepsin K is a lysosomal cysteine protease present in activated osteoclasts for degrading collagen and other matrix proteins during bone resorption. 127 MIV-711 is a potent selective cathepsin K inhibitor that revealed reduced subchondral bone loss and cartilage damage in animal models. 128 In a 26-week phase II human trial in knee OA (n = 244), significant reductions in bone and cartilage OA progression were detected but with no symptomatic benefits. Skin disorders were more common in the active groups (100 mg/day: 7.3%; 200 mg/day: 12.2%, placebo: 2.5%). 113
Matrix extracellular phosphoglycoprotein
Matrix extracellular phosphoglycoprotein (MEPE) is largely detected in normal osteocytes and downregulated in OA. 129 TPX-100 is a synthetic 23mer peptide fragment of MEPE (AC-100, region 242-264). 130 Intra-articular injections of TPX-100 stimulate articular cartilage proliferation in goats. 115 A phase II study involving 93 participants with bilateral patellofemoral OA, 4 weekly injections of 200 mg TPX-100, demonstrated a significant difference in pain when ascending and descending stairs at 12 months but no structural benefits on quantitative MRI, 114 perhaps due to the small sample size. On a post hoc clinical ‘responder’ analysis for participants having bilateral tibiofemoral cartilage defects (73%), more TPX-100-treated knees met the responder criteria than placebo-exposed knees. 131 Using a retrospective MRI study, TPX-100 (n = 78) demonstrated a statistically significant decrease in pathologic bone shape change in the femur and associations with less tibiofemoral cartilage loss. 115
Synovitis-driven endotype
XT-150
Due to a short half-life and low target concentrations in the joint when systemically administered, plasmid DNA-based technology was used to generate XT-150 as a long-acting human IL-10 variant. 132 It is well tolerated and provides a symptomatic improvement in canine OA model. 133 On exploratory analyses of the combined phase I studies by using a WOMAC (Western Ontario and McMaster Universities Osteoarthritis Index) pain responder analysis (30% reduction from baseline versus day 180), a significant difference in efficacy was observed for 150 µg XT-150 compared with the placebo (67% versus 21%). 123
Currently, its efficacy is being evaluated in a phase II clinical trial for knee OA (NCT04124042). It was reported that 215 subjects have already been enrolled out of planned enrolment of 270 subjects as of February 2021. 134
Diacerein
Diacerein inhibits the IL-1b system and related downstream pathways in mice. 135 Administration of diacerein 50 mg once per day for 1 month and twice daily thereafter (n = 140) in moderate and severe knee OA showed comparable efficacy in symptomatic improvement with celecoxib 200 mg once per day for 6 months (n = 148) despite more prevalence of GI side effects (10.2% versus 3.7%). The study was also limited as no structural outcomes were evaluated. 116 The EMA’s Pharmacovigilance Risk Assessment Committee recommended restrictions of its use to limit risks of severe diarrhoea and hepatotoxicity in 2014. 136 In a recent meta-analysis, diacerein was significantly related to GI disorders (OR: 2.53; 95% CI: 1.43 to 4.46) and renal disorders (OR: 3.16; 95% CI: 1.93 to 5.15) even when concomitant OA medications were not allowed. 137
Cartilage-driven endotype
Proteinase inhibitors
Collagenases such as MMPs and aggrecanase such as a disintegrin and metalloproteinases with thrombospondin motifs (ADAMTS) digest triple-helical type II collagen fibrils (collagenolysis) and aggrecan, the major proteoglycan in articular cartilage. 138 Due to severe adverse events such as musculoskeletal syndrome (arthralgia, myalgia, tendinitis) and GI disorders, further development of broad-spectrum MMP inhibitors such as PG-116800 has been terminated. 139
Although the involvement of MMP-13 in OA pathogenesis is suggested by preclinical studies, 140 MMP-13 inhibitors with superior selectivity profiles by occupying themselves deeper in the S1′ pocket have not reached the phase II clinical trials due to poor solubility, metabolic stability or bioavailability. 141
As ADAMTS-5 inhibition reduces joint damage in mice and human chondrocytes. 142 S201086/GLPG1972 has been developed as an inhibitor of ADAMTS-5 142 with an eightfold selectivity over ADAMTS-4 in preclinical studies. 143 Neither symptomatic nor disease-modifying benefits detected by MRI were found in a phase II study (Roccella study) for knee OA (n = 932) 117 despite the fact that the study design was optimized for capturing cartilage changes. 144 Phase I studies for an ADAMTS-5 nanobody (M6495) showed overall acceptable safety at single doses up to 300 mg for further clinical development. 145
Fibroblast growth factor 18
Sprifermin is a recombinant human fibroblast growth factor 18 (FGF18), derived from Escherichia coli expression system. 146 It increased the synthesis of extracellular matrix in animal studies. 147 In 2007, a First-in-Human (FiH) trial (NCT00911469) revealed its beneficial effects on the cartilage samples taken from 73 end-stage knee OA participants during KR surgery. 148 In 2008, a significant dose-dependent disease-modifying response was detected in prespecified secondary radiograph and MRI outcomes over 12 months but with no symptomatic benefits (n = 168).118,149,150
The third phase II clinical trial [FGF18 Osteoarthritis Randomized Trial with Administration of Repeated Doses (FORWARD) study] (NCT01919164) confirmed the previous findings at 2- and 3-year follow-ups, especially with the dose of 100 µg sprifermin (n = 549). 119 In post hoc analyses, location-independent reduction of cartilage loss was reported, 151 and a symptomatic benefit on WOMAC score at 3-year follow-up [−8.8 (−22.4, 4.9)] was observed in a ‘subgroup at risk’ with narrower medial or lateral minimum joint space width (mJSW) and higher WOMAC pain, identifying a potential target group for future sprifermin clinical trials. 152
Recently, the results of the 5-year FORWARD study (n = 494) demonstrated the maintenance of structure-protective effects of 2-year administration of sprifermin 100 µg every 6 months despite a treatment-free period of 3 years, with a good safety profile. 120 A recent meta-analysis pooling data from eight reports of three original trials confirmed its disease-modifying properties such as improvement in cartilage thickness, volume and morphology. 153 However, it failed to exhibit symptomatic improvement measured by WOMAC subscores. As a note, structure-protective effects may prevent or delay KOA patients from reaching levels of debilitating pain in the long term despite lack of initial symptomatic improvement.
Transforming growth factor-β
Transforming growth factor-β (TGF-β) is involved in the extracellular matrix protein synthesis 154 and regulation of subchondral bone remodelling. 155 TissueGene-C (TG-C) is a retrovirally transduced agent for stimulating TGF-beta1 transcription (hChonJb#7 cells). 121 Its cartilage-regeneration potential was confirmed in a recent study in OA rat model. 156
In a phase II trial, the IA TG-C administration (n = 57) reduced the cartilage damage and inflammation markers compared with the placebo (n = 29) over 12 months. 121 In a 2017 poster abstract, a single IA administration of the TG-C showed symptomatic benefits but inconclusive effects on cartilage measures at 12-month follow-up (n = 156). 157 These findings were confirmed by another study in 102 OA participants. 122 A 1-year phase III trial revealed a reduction in pain of 25% with TG-C treatment compared with 10% with the placebo group and no significant structural benefits on the JSN and MRI outcomes as the secondary endpoints (n = 163), 123 despite exhibiting a clear trend towards slower bone area progression in the TG-C group. The failure to meet the structural endpoints may be perhaps due to the limited image quality and statistical power. The temporary clinical hold in April 2019 over the concerns of the potential mislabelling of ingredients was lifted in April 2020. 158 An analysis of observational long-term safety follow-up data showed no evidence of its association with increased risk of cancer over an average 10 years. 159 There are two ongoing pivotal phase III trials (NCT03203330, NCT03291470).
Wnt signalling inhibitors
Increased Wnt signalling in the chondrocytes, subchondral bone and synovium leads to cartilage damage, bone sclerosis and production of MMPs, respectively.160–162 Lorecivivint (SM04690) is a small-molecule CLK/DYRK1A Wnt signalling inhibitor that reduced cartilage damage in preclinical models. 163 In a 52-week, phase IIa trial in 455 participants with knee OA, any Lorecivivint dose group did not meet the primary endpoint, significant improvement in the WOMAC pain score at week 13 in comparison with the placebo group. However, in the post hoc analyses in subgroup of participants with unilateral symptomatic knee OA (n = 164) or unilateral symptomatic knee OA but without widespread pain (n = 128), administration of 0.07 mg resulted in significant symptomatic and radiographic improvements at 52 weeks, 124 identifying a potential responsive target phenotype. In another phase IIb clinical trial (NCT03122860) (n = 700), the lowest optimal dose was determined as 0.07 mg, 125 supporting the previous findings. The analysis of the combined data of these two trials showed a favourable safety profile (848 = Lorecivivint-treated and 360 = control subjects). 164 There are several ongoing trials on the https://clinicaltrials.gov/: two small phase II (NCT03727022, NCT03706521) and three phase III (NCT03928184, NCT04385303, NCT04520607) trials.
UBX0101
Recently, senescence (termination of cell division) caused by proinflammatory cytokines and dysfunction of neighbouring cells165,166 is focused on the pathogenesis of ageing-related OA. 167 As a senotherapeutic, UBX0101 is a p53/MDM2 interaction inhibitor and promoted chondrogenesis in animal models. 168 Although it showed encouraging results in a phase I study (n = 48), 169 a 12-week phase II study failed to reveal symptomatic improvement (n = 183), 126 leading to the termination of a long-term follow-up observational study. As senolytics appear to be cell type specific, a single senolytic drug may not be able to eliminate all types of senescent cells, 170 attributing to possible reasons for failures of single-drug RCT to meet the trial endpoints. Future research to improve the specificity and potency of senolytics by using delivery system such as galactose conjugation examines the efficacy and safety profiles of intermittent versus continuous administration, and a combination of senolytic agents should be considered. 171
LNA043
LNA043 is a modified human angiopoietin-like 3 (ANGPTL3) protein. A single IA injection of 20 mg LNA043 had anabolic effects on the cartilage via forming hyaline-like tissue detected on the high-field 7-T MRI. 172 In a dose-finding phase I study (NCT02491281), it showed dose-dependent modulation of cartilage-repair genes in the damaged cartilage tissue acquired from knee OA patients scheduled for total knee replacement (TKR), with a favourable safety profile (n = 28). LNA043 possessed rapid penetration predominantly into the damaged cartilage within 2 h after IA injection and was undetectable in the articular cartilage or synovial fluid 7 days post injection. 173
A phase IIa/b study (NCT03275064) is ongoing. In the analysis of the phase IIa part of this clinical trial, significant regeneration of damaged cartilage was detected up to week 28 on the femoral lesions but not on the patellar lesions after 4 weekly IA injections of 20 mg LNA043 in participants with a partial thickness cartilage lesion. There was higher incidence of joint swelling (9.3% versus 0%) and arthralgia (11.6% versus 6.7%) in the LNA043 group (n = 43), compared with the placebo (n = 15). 172 On the https://www.clinicaltrialsregister.eu/, a recent record for a 5-year clinical trial (ONWARDS) was found (EudraCT number: 2020-004897-22). It was granted FDA fast track designation (a process used to expedite the progress and review of new drugs which demonstrates the potential by theoretical or mechanistic rationale to address unmet medical needs 174 ) for knee OA in September 2021. 175
LRX712
LRX712 stimulates differentiation of chondroprogenitor cells to generate new extracellular matrix. 176 In a phase I clinical trial (NCT03355196) completed in 2019, it had a favourable safety profile with most of the adverse events being the injection site pain and swelling in the active group (n = 28) compared with the placebo (n = 14) (up to 75% in the highest dose versus 7%). 177 A phase II study is underway to examine its cartilage morphometrics detected by 7-T MRI and estimated to be completed in early 2024 (NCT04097379).
Perspectives
Recent failures and potential reasons (DMARDs, GLPG1972)
Despite the immense success of DMARDs in the remission of disease activity in inflammatory arthritides such as RA, 83 neither conventional (i.e. MTX, 37 HCQ 40 ) nor biological DMARDs (i.e. etanercept, 96 tocilizumab 42 ) in recent clinical trials have brought success in improving either symptomatic or structural benefits or both in the case of predominantly inflammatory OA endotypes. These failures may be attributable to the involvement of more complex interactions among various inflammatory cytokines in developing this endotype, 1 an insufficient number of participants, too short follow-up duration, recruiting participants with low disease activity or insufficient inflammatory activity to capture the symptomatic or structure-modifying efficacy of these medications. 9 However, there is no existing consensus on the threshold of optimal follow-up duration which is sufficient for detecting DMOAD effects 13 and the recent exploratory analysis of Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) trial suggests that to see the benefits for modulating inflammation may take longer (averaged follow-up duration of 3.7 years). 178 One of the other limitations may be due to the disease definition itself as the commonly used inclusion criteria in DMOAD trials [i.e. the American College of Rheumatology (ACR) criteria and radiographic criteria such as KL grade 2 or 3 on plain radiograph] may miss an early responsive window period where the best disease-modifying opportunities may be available for diagnosis and initiation of DMOAD therapy 13 as evidenced in paradigm shift in RA management in the last decade. 179 Therefore, a consensus definition for identifying either an ‘early OA’ or the pre-stages of OA for the purpose of DMOAD clinical trials may improve the chances of success in future trials.
In the case of proteinase inhibitors (PIs) such as GLPG1972, a major issue in targeted therapies is the high degree of conservation of proteinases in the sequence and structure of the active site, causing undesirable cross-inactivation of multiple proteinases and off-target effects when systemically administered. 180 As an example, while inhibition of the aggrecan degradation is directed to the articular cartilage, 181 simultaneous inhibition in tendon and aorta leads to decreased mechanical properties 182 and aortic dissection and rupture. 183 Therefore, linkage of the molecular target with the disease and fulfilments of the target agent with the attributes of receptor or functional selectivity, specificity and potency are crucial 184 potentially benefitted by long-acting intra-articular administration.
Recent insights/successes (sprifermin, Lorecivivint)
Sprifermin seems to have structure-modifying capability as well as long-term maintenance of its effects, based on the 5-year phase II data. One major strength of this study is using quantitative MRI as the primary outcome, resulting in an observed difference in MRI-detected cartilage thickness of significant results of pre-planned and post-hoc the medial femorotibial compartment while there was no difference in change of mJSW in the medial compartment. 119 Although the study did not meet its secondary endpoint, change in WOMAC score, it was not primarily designed as a pain trial with 90% in each treatment group taking additional pain medications. In addition, the participants were heterogeneous (32% with WOMAC pain subscale score <40/100) and relatively high mJSW (50% >3.7 mm) at baseline. IA saline injections were used as the placebo, masking any symptomatic benefits.
The post hoc analysis in a more homogeneous OA patient subgroup provided further insight that a symptomatic response to treatment is more likely in homogeneous symptomatic subgroups with rapid progression of the disease. 152 The time lag of symptomatic improvement translated from structural improvement is more than 2 years which may explain the failure of some clinical trials with shorter duration or with heterogeneous patient populations. In addition, a trial population with low mJSW and high WOMAC pain at baseline may be more effective to fulfil the definition of a DMOAD.
Although the Lorecivivint clinical trial did not meet the primary endpoint of WOMAC score, 124 the significant results of pre-planned and post hoc analyses in participants with unilateral symptoms of knee OA provided further insight for designing future clinical trials. 125 As the ability of a participant to discriminate pain between target and non-target knees is crucial, selection of participants can be limited to the unilateral OA symptoms, based on pain NRS (Numerical Rating Scale) cut-off points between knees during screening (i.e. the contralateral knee must have had a daily average NRS intensity score <4 for 4 of 7 days). In this way, a potential symptomatic improvement could be more clearly delineated.
Novel methodologies that may enhance success (placebo response tools, drug delivery system)
For obtaining regulatory approval, RCTs are the ‘gold standard’ to prove the efficacy and safety of a medical intervention. In an RCT, a placebo has to be administered in the control group to compare with the active agents in terms of benefits and adverse events for evidence generation. 185 As observed in many of the DMOAD RCT discussed above, the control group showed symptomatic improvement in the longitudinal follow-ups. A recent meta-analysis showed that the placebo effects of IA saline at 6-month follow-up [−13.4 (−21.7, −5.1) on a 0–100 VAS pain score and −10.1 (−12.2,-8.0) WOMAC function subscore] are higher than the ‘minimal clinically important difference’ (13.7/100 for VAS pain score and 4.6/68 for WOMAC function score). 186 In addition, the placebo response tends to increase in proportion to the effect size of the active treatment. 187 This high long-lasting placebo response seems a challenge for designing a clinical trial and may mask the efficacy of the potential DMOADs. 14
The use of needles or injections gives rise to greater placebo (contextual) effects [proportion attributable to contextual effects (PCE) = 0.81, 95% CI: 0.75 to 0.88], compared with oral medications. 188 To increase the accuracy of the IA drugs, ultrasound guidance is frequently used in DMOAD RCT. The use of such a sophisticated imaging tool may provide greater placebo response 189 as observed as high placebo response in invasive procedures. 190 Therefore, the placebo effects of IA saline should be accounted for in planning the trial design when pain and function endpoints are used as the primary measures.13,191 Furthermore, placebo response tools for predicting the placebo response for a particular intervention in a specific disease are being developed. 192
Due to the higher prevalence of slowly progressive OA in the fragile elderly with multiple comorbidities, 7 systemic administration could cause off-target effects and undesirable systemic effects over the long term. On the contrary, local therapy such as the IA administration will directly reach the targeted organ and requires a lower dosage. 193 Still, the short half-life/residence times of the agents in the joints is a major barrier to progress leading to frequent IA injections and burden for both participants and medical practitioners. Several drug delivery systems (DDS) which are capable of controlled and/or sustained drug release are being developed to prolong the residence time in the joint. 194 New smart drug delivery strategies, using nanoparticle, microparticle and hydrogel methods, may maximize the efficacy and safety of intra-articular agents. 195
Steps underway to enhance approval (Foundation for NIH qualification efforts, accelerated approval regulations)
Due to the FDA’s formal recognition of OA as a serious disease, utilization of surrogate outcome measures becomes feasible to submit the findings to the regulatory bodies for accelerated approval regulations. However, there remain two challenges ahead: (1) selection and evaluation of relevant surrogate outcome measures, and (2) appropriate designs for postmarketing confirmatory studies. The Foundation for NIH (FNIH) OA Biomarkers Consortium initiative was established to address the first challenge. 196 Two major study design scenarios were put forward to address the second challenge: (1) prospective trial continuation, which permits all participants on initial drug allocation to continue into the postmarketing approval trial until reaching a failure threshold; and (2) separate postmarketing approval study which recruits different study population to be treated with active medication only. 197
Conclusion
With the increasing combined trends of aeging, and rising epidemic of obesity, OA will lead to a major public health issue in the coming decades but with an immense unmet need for effective and safe therapies despite massive efforts and investments in R&D pipelines. To cut off the escalating costs, drug repurposing strategies are being used for finding a DMOAD but without success in OA so far. Recently, a few investigational drugs revealed promising findings in late clinical trials, paving the way for selecting a population at risk and developing more novel trial designs. Lessons learned from past failed clinical trials and insights gained through the successful progression of drug development phases, utilization of novel methodologies/techniques such as placebo response tools/DDS and steps to overcome barriers to regulatory hurdles will facilitate the emergence of the first DMOAD to fulfil the unmet need of patients with OA.
Footnotes
Acknowledgements
D.J.H. is supported by an National Health and Medical Research Council (NHMRC) Investigator Grant. W.M.O. was supported by the Presidential Scholarship of Myanmar for his PhD course.
Author contribution(s)
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: D.J.H. provides consulting advice on scientific advisory boards for Pfizer, Lilly, TLCBio, Novartis, Tissuegene and Biobone. W.M.O. has no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.