
Abstract
Axial spondyloarthritis (axSpA) is a chronic inflammatory rheumatic disease characterized by inflammation and new bone formation in the axial skeleton. AxSpA is considered a spectrum of disease that includes two subtypes identified by the Assessment in SpondyloArthritis International Society classification criteria, namely, radiographic (r-axSpA usually referred to as ankylosing spondylitis) and non-radiographic axSpA (nr-axSpA). Although the burden of disease appears similar between the two classified subtypes, the degree of inflammation, as assessed by magnetic resonance imaging and C-reactive protein, and the degree of new bone formation are significantly higher in r-axSpA than in nr-axSpA. Nevertheless, axSpA is considered one disease with different courses. International guidelines for the management of axSpA have outlined treatment goals focused on control of signs and symptoms, inflammation, prevention of progressive structural damage, preservation of physical function, normalization of social participation and improvement of quality of life. The pathogenesis of axSpA has not been completely elucidated to date. A strong link between human leukocyte antigen B27 and axSpA, however, has been identified, and the success of anti-tumour necrosis factor and anti-interleukin (IL)-17A therapy has highlighted some of the key pro-inflammatory cytokines involved. The anti-IL-17A monoclonal antibody secukinumab is approved for the treatment of ankylosing spondylitis and nr-axSpA in the European Union and United States. In this narrative review, we discuss data for secukinumab in axSpA from randomized controlled trials, including MEASURE trials in AS and PREVENT in nr-axSpA, and real-world evidence.
Keywords
Introduction
Axial spondyloarthritis (axSpA) is an inflammatory rheumatic disease characterized by inflammation and bone formation in the axial skeleton where it causes inflammatory back pain.1,2 Usually starting in the sacroiliac joints (SIJ) during the second to third decade of life, axSpA frequently spreads to the spine. These inflammatory changes in the axial skeleton are well visualized by magnetic resonance imaging (MRI). A consequence of inflammation is the growth of syndesmophytes and the possible development of ankyloses in the spine that can be detected on conventional radiographs. 3 The latter has led to the term ankylosing spondylitis (AS), which has been the major heading in textbooks for decades and is still frequently used.
Peripheral joint symptoms of axSpA, such as arthritis and enthesitis, occur in many sites, most commonly the Achilles tendon and the plantar fascia.1,4 In addition to these characteristics, approximately 40–45% of patients with axSpA experience extra-musculoskeletal manifestations, such as anterior uveitis and psoriasis. 5 Comorbidities are also common in patients with axSpA, with cardiovascular disease and osteoporosis often being observed. 6
Originally, in accordance with 1984 New York classification criteria, a diagnosis of axSpA was rarely made if there was no evidence of radiographic structural damage in the SIJ. 7 Since then, there has been successful introduction of MRI for the detection of inflammation in the SIJ and the spine, introduction of guidelines relating to drug approvals for AS and the publication of the Assessment of SpondyloArthritis International Society (ASAS) classification criteria in 2009. 7 The axSpA subtype known as non-radiographic (nr)-axSpA is defined by the absence of definite radiographic structural changes in the SIJ, while the corresponding subtype radiographic (r-)axSpA is largely equivalent to the classical AS. 8 Hence, axSpA can be considered one spectrum of disease that includes nr-axSpA and r-axSpA. 8
The disease burden is largely similar in r-axSpA and nr-axSpA, but there are some differences between the two subtypes. The nr-axSpA patient population generally includes more females, less C-reactive protein (CRP) and MRI changes and only minor radiographic progression over time as detected by the modified Stoke Ankylosing Spondylitis Spine Score (mSASSS). 9 The burden of disease in axSpA can be understood and quantified following the introduction of the ASAS Health Index (ASAS-HI) (Supplementary Table 1).10,11 A study found that patients with axSpA (disease duration > 10 years) reported pain as the most important feature of their disease, followed by sleep disturbance and exhaustion. 12 Taken together, many aspects of patients’ daily lives are affected by the disease; this includes depression, psychological distress, reduced physical function and work disability.12,13
Treatment goals for axSpA include improving long-term health-related quality of life through control of symptoms and inflammation, prevention of progressive structural damage, preservation of function and normalization of social participation.1,14 The ASAS-European League Against Rheumatism (ASAS-EULAR) and American College of Rheumatology-Spondylitis Association of America-Spondyloarthritis Research and Treatment Network (ACR-SAA-SPARTAN) guidelines, two international guidelines commonly used in the management of patients with axSpA, both recommend non-steroidal anti-inflammatory drugs (NSAIDs) as the first-line treatment for axSpA.1,15 NSAIDs initially relieve symptoms in 70–80% of patients with axSpA; only one third of patients, however, achieve adequate disease control with NSAIDs and the impact of these therapies on preventing radiographic progression is unclear. 16 For decades, NSAIDs and physiotherapy were the only treatments available for patients with axSpA; hence, available treatment options for patients refractory to conventional therapy were limited. 17
The advent of biological disease-modifying anti-rheumatic drugs (bDMARDs) at the start of the 21st century revolutionized the axSpA treatment landscape. 18 The first bDMARDs available for treating axSpA blocked the tumour necrosis factor (TNF)-alpha inflammatory pathway as part of their mechanism of action. Anti-TNF therapies have demonstrated efficacy in reducing pain and stiffness across the axSpA spectrum. 19 Despite this, 20–30% of patients with axSpA do not respond adequately to TNF inhibitors, resulting in the need for new therapies with alternative mechanisms of action. 19 Interleukin (IL)-17A, a mediator in inflammatory pathways, has been identified as a key cytokine in the pathogenesis of axSpA. This has led to the development of IL-17A inhibitors for the treatment of axSpA. 20
Secukinumab
The discovery of T helper type 17 (Th17) cells was an important advance in understanding the pathogenesis of inflammatory diseases. 21 This was soon followed by the identification of the pro-inflammatory cytokine IL-17A, an effector cytokine of Th17 cells. 22 Both adaptive immune cells, such as Th17, and innate immune cells, such as ILC3, have been implicated in IL-17A production. 20 Upon binding to its receptor, IL-17A upregulates inflammatory gene expression by stabilizing pro-inflammatory cytokine mRNA and inducing de novo gene transcription. 20 IL-17A has been shown to play a key role in the pathogenesis of psoriasis (PsO), psoriatic arthritis (PsA) and axSpA. As a result, biologics targeting IL-17A signalling pathways have been developed for treating these diseases. 23 At present, there are two anti-IL-17A biologics approved for the treatment of axSpA: secukinumab and ixekizumab. 24 Further anti-IL-17 biologics, such as brodalumab and bimekizumab, are being evaluated in clinical trials in patients with axSpA; these therapies, however, are still in clinical development and are not currently indicated in this patient population.25,26
Secukinumab is a fully human IgG1/κ monoclonal antibody that selectively binds to IL-17A and inhibits its interaction with the IL-17 receptor, reducing IL-17A-mediated contributions to inflammatory diseases. 27 In 2016, secukinumab became the first IL-17A inhibitor, and the first non-anti-TNF biologic therapy, to be approved for the treatment of adults with active r-axSpA who have responded inadequately to NSAIDs. 28 In 2020, secukinumab was approved for the treatment of adults with nr-axSpA with objective signs of inflammation who have responded inadequately to NSAIDs. 24 In the European Union (EU), the recommended dose of secukinumab for patients with axSpA is 150 mg by subcutaneous injection, with loading doses administered at weeks 0, 1, 2, 3 and 4, followed by subsequent doses administered every 4 weeks. 27 In contrast, in the United States (US), secukinumab 150 mg can be administered with or without the loading dose. 29 Based on clinical response, the dose can be increased to 300 mg for patients with r-axSpA.27,29 Indications and dosages for secukinumab in patients with axSpA are based on results of the MEASURE clinical trial programme (r-axSpA) and the PREVENT trial (nr-axSpA) (Table 1). Patients who were anti-TNF-naïve or had an inadequate response to or intolerance of previous anti-TNF therapy were enrolled across studies.23,30–32 This narrative review will primarily focus on r-axSpA data from MEASURE 2, due to the secukinumab dosing regimen approved by the European Medicines Agency (EMA); key data from MEASURE 1 and 3 are also highlighted where appropriate.

ASAS, Assessment of SpondyloArthritis international Society; IV, intravenous; LD, loading dose; NL, non-loading dose; PBO, placebo; QW, once a week; Q4W, once every 4 weeks; SC, subcutaneous; SEC, secukinumab; TNF, tumour necrosis factor.
PREVENT was the first randomized, placebo-controlled phase III trial studying the efficacy and safety of secukinumab in patients with nr-axSpA. Patients who were anti-TNF-naïve or had an inadequate response to previous anti-TNF therapy were included in the trial, although approximately 90% of patients were anti-TNF-naïve at baseline. 33
Here, we review secukinumab data from RCTs and real-world evidence (RWE) to evaluate how this IL-17A inhibitor addresses treatment goals and reduces disease burden for patients with axSpA.
Signs and symptoms
ASAS response criteria are used to assess improvements in signs and symptoms of axSpA and as such are a composite measure that encompass four domains: pain, inflammation, function and patient global assessment of disease activity. 34 These criteria are commonly used in clinical trials to assess improvement and response to treatment in axSpA. 35 The primary endpoint of MEASURE 2 was the proportion of patients who achieved ASAS20 at week 16 in the overall population. 23 ASAS20 is defined as an improvement of greater than or equal to 20% in three of four domains of ASAS response and a minimum of one unit on a scale from 0 to 10. In the remaining domain, there should be no worsening of 20% and a minimum of 1 unit, on a 0–10 scale. 35 The primary endpoint of MEASURE 2 was met, with significantly higher proportions of patients achieving ASAS20 versus placebo; improvements were sustained through to 5 years (Figure 1).23,36
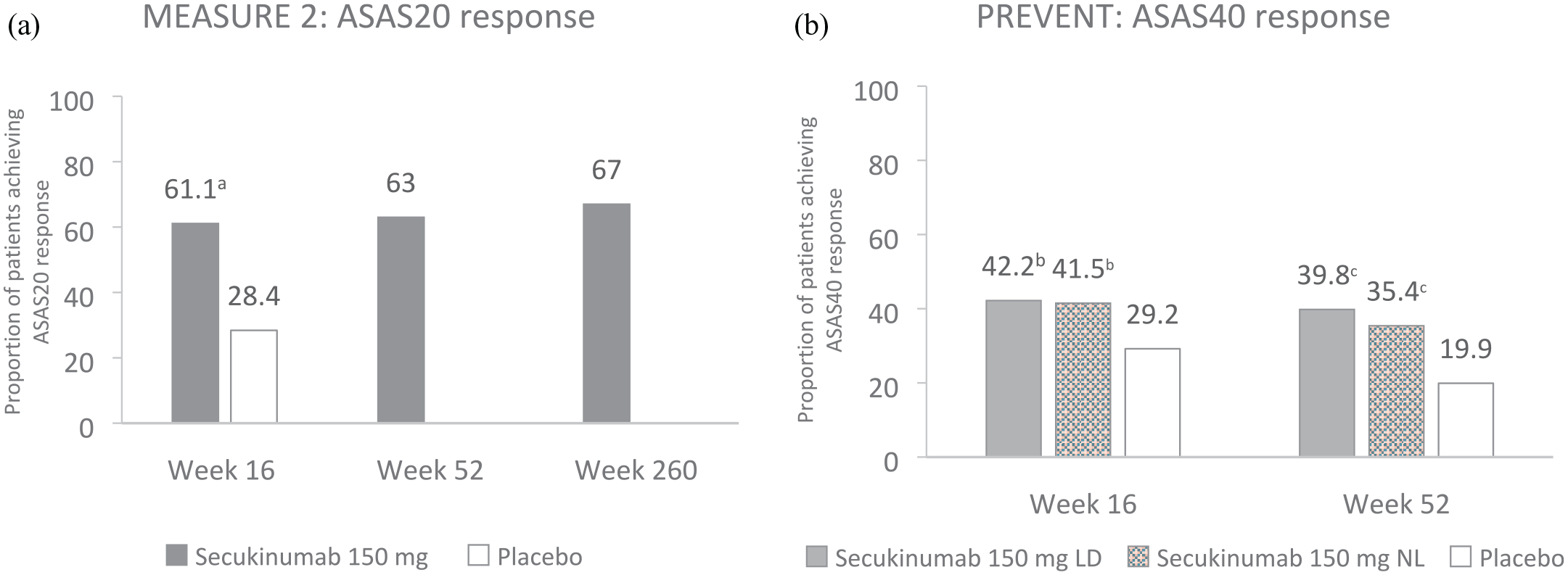
Summary of (a) ASAS20 response in MEASURE 2 and (b) ASAS40 response in PREVENT at week 16, week 52 and week 260 (MEASURE 2 only).23,33,36 MEASURE 2: Data presented using non-responder imputation at weeks 16 and 52 in the overall population of patients with r-axSpA; presented as observed at week 260 in the overall population. Week 16: secukinumab 150 mg, n = 72; placebo, n = 74. Week 52: secukinumab 150 mg, n = 72. Week 260: secukinumab 150 mg, n = 54. The primary endpoint of MEASURE 2 was the proportion of patients who achieved ASAS20 at week 16 in the overall population. PREVENT: Data presented using non-responder imputation through to week 52 in the TNFi-naïve population of patients with nr-axSpA. Secukinumab 150 mg LD, n = 164; secukinumab 150 mg NL, n = 166; placebo, n = 171. The co-primary endpoints of PREVENT were the proportion of patients in the anti-TNF-naïve population who achieved ASAS40 at weeks 16 (loading dose) and 52 (non-loading dose).
In PREVENT, co-primary endpoints were the proportion of patients in the anti-TNF-naïve population who achieved ASAS40 at weeks 16 (loading dose) and 52 (non-loading dose); these time points were selected to satisfy EU and US regulatory requirements, respectively. 33 ASAS40 is defined as an improvement of greater than or equal to 40% in three of four domains of ASAS response criteria (patient global, pain, function and inflammation) and a minimum of two units on a scale from 0 to 10. In the remaining domain, there should be no worsening of 20% and a minimum of 1 unit, on a 0–10 scale. 35 Both co-primary endpoints were met in PREVENT; significantly higher proportions of anti-TNF-naïve patients achieved an ASAS40 response with secukinumab 150 mg loading dose at week 16 and non-loading dose at week 52 versus placebo (Figure 1). 33
Pain
Spinal pain is a major contributor to the patient burden of axSpA, with 30–35% of patients reporting this as their most burdensome symptom. 37 Pain is often assessed using two standard questions relating to the amount of pain experienced during the day and night, either using a visual analogue scale (VAS) or a numeric rating scale (NRS). 35 Rapid and sustained improvements in pain have been observed with secukinumab in patients with r-axSpA. In MEASURE 2, improvements in overall spinal pain and nocturnal back pain were observed as early as week 1. 37 Patients receiving secukinumab achieved significant improvements in total spinal pain VAS score versus placebo (–29.0 versus –11.4, p < 0.0001) at week 16. 37 Significant improvements were also observed at week 16 in nocturnal back pain VAS score for secukinumab 150 mg versus placebo (–30.3 versus –10.1, p < 0.0001). 37 These reductions were sustained over 5 years of treatment. 38 Higher proportions of patients receiving secukinumab 150 mg in MEASURE 2 met minimal clinically important difference (MCID) criteria for improvements in nocturnal back pain compared with placebo at week 16 in the overall population (Figure 2). Reductions in pain were observed regardless of previous anti-TNF therapy; the magnitude of improvement, however, was numerically greater in patients who were anti-TNF-naïve compared with anti-TNF inadequate responders. 39

Proportions of patients meeting MCID criteria in MEASURE 2 and PREVENT for (a) nocturnal back pain, (b) FACIT-F and (c) morning stiffness.38–40 MEASURE 2: Observed data are presented through week 260. Secukinumab 150 mg, n = 72 and placebo, n = 74; n = 67 and n = 54 at week 16 and 260, respectively, in the secukinumab 150 mg group; n = 64 at week 16 in the placebo group (overall population). Data at week 16 are not statistically significant as no statistical analysis was performed. 38 PREVENT: Data presented using non-responder imputation at week 16; presented as observed at week 52. Secukinumab 150 mg loading dose, N = 164 and placebo, N = 171 (anti-TNF-naïve population). 33 Pain and morning stiffness at week 52: secukinumab 150 mg loading dose, n = 139; patients switched to open-label secukinumab 150 mg, n = 67. Fatigue at week 52: secukinumab 150 mg loading dose, n = 146; patients switched to open-label secukinumab 150 mg, n = 69. 40 MCID definition: nocturnal back pain, defined as an improvement from baseline of ⩾50%; FACIT-F: defined as an improvement from baseline of ⩾4 points; morning stiffness: defined as an improvement from baseline in BASDAI average of ⩾22.5%. 40
Similarly, in PREVENT, secukinumab significantly reduced nocturnal back pain in anti-TNF-naïve patients with nr-axSpA. Mean changes from baseline of −30.93 and −31.91 points were achieved with secukinumab 150 mg loading dose and non-loading dose, respectively, versus –20.27 for placebo at week 16 (p < 0.001). Improvements were achieved as early as week 4 and sustained to week 52. 40 Significantly higher proportions of patients receiving secukinumab 150 mg met MCID criteria for improvements in nocturnal back pain compared with placebo at week 16 (Figure 2).
Further evidence for the efficacy of secukinumab in reducing spinal pain has been demonstrated in SKIPPAIN, the first randomized controlled trial (RCT) involving a bDMARD with spinal pain as the primary endpoint as early as week 8 in patients across the axSpA spectrum (both r-axSpA and nr-axSpA). The primary endpoint was the proportion of patients achieving an average spinal pain score of less than 4 on a 0–10 NRS with secukinumab versus placebo at week 8; this was achieved following significant improvements in average spinal pain score with secukinumab versus placebo (31.9% versus 20.0%, p < 0.05). 41
Fatigue
Fatigue remains an unmet need in axSpA, with approximately 50–65% of patients reporting fatigue as their most troubling symptom. 37 Secukinumab has been shown to improve fatigue in axSpA through rapid and sustained improvements in Functional Assessment of Chronic Illness Therapy–Fatigue (FACIT-F) scores. In MEASURE 2, least square mean improvements in FACIT-F total scores from baseline were significantly higher with secukinumab 150 mg versus placebo at week 16 (8.10 versus 3.27, p < 0.01). 42 Improvements in fatigue were sustained through to 5 years and were consistent in both anti-TNF-naïve and anti-TNF inadequate responder populations.38,39
In addition to FACIT-F score, fatigue in axSpA may be measured using question 1 of the Bath Ankylosing Spondylitis Disease Activity Index (BASDAI). 37 In MEASURE 2, patients with r-axSpA receiving secukinumab 150 mg demonstrated significant improvements in fatigue at week 16 versus placebo, as demonstrated by the mean change in BASDAI question 1 score from baseline (–2.0 versus –1.0, respectively, p < 0.01). 37 Improvements were sustained through to 5 years.
For anti-TNF-naïve patients in PREVENT, significant improvements in mean change from baseline in FACIT-F score were achieved with secukinumab 150 mg loading dose (9.10, p < 0.001) and non-loading dose (8.61, p < 0.01) versus placebo (5.25) at week 16. 40 In both MEASURE 2 and PREVENT, rapid and sustained improvements in fatigue were achieved with secukinumab 150 mg, demonstrated by the proportions of patients achieving MCID criteria for FACIT-F (Figure 2).38,40
Stiffness
Spinal stiffness is a debilitating clinical symptom associated with axSpA. Approximately up to 76% of patients with axSpA report moderate-to-severe spinal stiffness throughout the day.43,44 Patients with stiffness may experience sleep disturbance due to discomfort, further affecting their quality of life. 45 Morning stiffness is assessed by BASDAI questions 5 and 6. 46 Observed data from MEASURE 2 demonstrate that improvements in morning stiffness were achieved with secukinumab 150 mg versus placebo at week 16, as measured by mean changes from baseline in average BASDAI score (–2.46 versus –0.86). 38 Improvements were observed as early as week 4 and sustained through to 5 years. 38 Reductions in morning stiffness were observed in patients receiving secukinumab 150 mg, regardless of previous anti-TNF therapy. 39 In PREVENT, rapid and sustained improvements in morning stiffness were observed with secukinumab 150 mg. Improvements in morning stiffness, measured as mean change from baseline, were significantly higher with secukinumab 150 mg loading dose (–3.30, p < 0.001) and non-loading dose (–3.34, p < 0.001) groups versus the placebo (–2.20) group at weeks 4 and 16 in the anti-TNF-naïve population. 40 Improvements were observed as early as week 4 and were sustained through to 52 weeks. 40 The proportion of patients meeting MCID criteria for morning stiffness were higher with secukinumab 150 mg versus placebo in MEASURE 2 and PREVENT (Figure 2).39,40
Disease activity
Persistent disease activity leads to poor patient outcomes; hence, it is important for patients with axSpA to achieve low disease activity, or remission, wherever possible. 47 Secukinumab 150 mg allowed patients to achieve partial remission across the axSpA spectrum in MEASURE and PREVENT, through fulfilment of ASAS partial remission (ASAS-PR) criteria. At week 16 in MEASURE 2, a numerically higher proportion of patients achieved ASAS-PR versus placebo (14% versus 4%). 23 After 5 years of treatment, 25% of secukinumab-treated patients achieved ASAS-PR. 36 In PREVENT, significantly more patients achieved ASAS-PR at week 16 with the loading (21.6% versus 7.0%, p < 0.0001) and non-loading (21.2% versus 7.0%, p = 0.0001) doses of secukinumab 150 mg versus placebo. 33
Data from MEASURE 3 demonstrated the efficacy of secukinumab 300 mg in reducing disease activity in patients with r-axSpA. 30 In the overall population, ASAS-PR was achieved by 21.1% of patients receiving secukinumab 300 mg, compared with 9.5% of patients receiving secukinumab 150 mg at week 16. 30 An increased response in the 300 versus 150 mg arm was observed through 3 years (28.3% versus 15.9%). 48 These data support the current recommendation for dose escalation from secukinumab 150–300 mg in patients with r-axSpA who demonstrate an inadequate clinical response with the lower dose.30,48
BASDAI is a patient-reported assessment of disease activity that incorporates symptoms of fatigue, back pain, peripheral joint pain and swelling, localized tenderness and morning stiffness. 46 Patients with r-axSpA in MEASURE 2 achieved a significantly greater mean change in BASDAI score from baseline with secukinumab 150 mg versus placebo (–2.19 versus –0.85, p < 0.001), which was sustained through to 5 years.23,36 Disease activity in axSpA may also be measured using the Ankylosing Spondylitis Disease Activity Score C-reactive protein inactive disease (ASDAS-CRP ID), describing a state of very low disease activity with an ASDAS score of less than 1.3. 49 In MEASURE 2, 21% of patients met ASDAS-CRP ID through to 5 years. 36 In PREVENT, 16% and 24% of patients in the secukinumab 150 mg loading and non-loading dose groups, respectively, achieved inactive disease up to 52 weeks. 33 High placebo responses for disease activity may be attained in clinical studies, often due to the subjective nature of endpoints such as ASAS20 and ASAS40. 33 In comparison, stringent disease activity endpoints such as ASDAS-CRP ID and ASAS-PR ensure that placebo responses remain low. 33
Function
Improving and preserving physical function is one of the main goals of axSpA treatment. 1 AxSpA has a major impact on physical function, impairing the ability of patients to carry out daily activities. 1 The Bath Ankylosing Spondylitis Functional Index (BASFI) is the most widely used functional index for assessment of axSpA. 35 Data from MEASURE 2 demonstrate significant improvements in function at week 16 with secukinumab 150 mg versus placebo (–2.15 versus –0.37, p < 0.0001); this was sustained through to 5 years.36,50 Similar improvements in function were observed with secukinumab 150 mg loading dose in patients with nr-axSpA, as observed by the change from baseline in BASFI score at week 16 versus placebo (−1.75 versus −1.01, p = 0.0041) in PREVENT, sustained through 52 weeks. 33 Following recommendations from ASAS-EULAR for exercise and physical therapy in patients with axSpA, the addition of physical activity could likely enhance these BASFI response rates. 1
Health-related quality of life
While many aspects of axSpA contribute to disease burden, the primary goal of axSpA is normalization of health-related quality of life.1,51 The Ankylosing Spondylitis Quality of Life Questionnaire (ASQoL) is a disease-specific instrument that quantifies the impact of axSpA on patients’ quality of life. 51 Secukinumab has been shown to improve ASQoL scores across the axSpA spectrum. In MEASURE 2, a mean change from baseline in ASQoL of −4.00 was achieved with secukinumab 150 mg versus −1.37 with placebo in the overall population at week 16 (p < 0.01). Improvement in ASQoL score was sustained through 1 year.23,52 In PREVENT, ASQoL scores were significantly improved with secukinumab 150 mg loading dose (−3.45, p = 0.0008) and non-loading dose (−3.62, p = 0.0002) versus placebo (−1.84) at week 16; improvements were sustained through 52 weeks. 33
Work productivity and activity impairment
The ability to work has a positive impact on psychosocial health; this is, however, impaired by musculoskeletal disorders such as axSpA. 53 Up to 30% of patients with axSpA are affected by work disability, and patients with axSpA are three times more likely to withdraw from work compared with those without axSpA, particularly from physically demanding jobs. 53 Presenteeism (productivity loss due to ill health) and work impairment are often experienced by patients with axSpA, which in turn leads to absenteeism (being absent from work due to ill health) and job loss. 53 Work Productivity and Activity Impairment (WPAI) is an instrument commonly used to measure work productivity in patients and includes questions to assess absenteeism, presenteeism and overall work productivity impairment. 54 In MEASURE 2, patients with r-axSpA receiving secukinumab 150 mg demonstrated clinically meaningful changes from baseline in absenteeism (−4.9), presenteeism (−19.2) and overall work impairment (−21.3). 54 Improvements were observed in all patients, regardless of previous anti-TNF therapy. 54 At present, there are no work productivity or activity impairment data available for secukinumab-treated patients with nr-axSpA.
Structural damage
One of the main treatment goals of axSpA is preventing structural damage, which usually begins with erosions in the SIJ and leads to bridging and subsequent ankylosis.1,55 New bone formation is a key process in the development of syndesmophytes, contributing to structural damage. 56 Structural damage identified on an X-ray is the key feature that differentiates r-axSpA from nr-axSpA. It is estimated that 10–40% of patients with nr-axSpA will progress to r-axSpA over 2–10 years, and greater than or equal to 50% of patients with nr-axSpA will likely progress to r-axSpA in their lifetime. 57 Risk factors for radiographic progression in nr-axSpA include HLA-B27 status, elevated CRP and active sacroiliitis on MRI. 57 Currently, the most frequently used instrument to assess radiographic damage is mSASSS. 35 In MEASURE 1, 78.9% of patients receiving secukinumab 150 or 75 mg, with a 10 mg/kg IV (intravenous) loading dose at baseline, had no radiographic progression (defined as a change in mSASSS from baseline of <2), which was sustained through 4 years. 58 At 2 years, 97.1% of patients in the overall population who had no syndesmophytes at baseline remained syndesmophyte-free (syndesmophytes were considered present if mSASSS was ⩾2 per specific affected vertebral unit). 59 At present, there are no studies investigating the effect of secukinumab on radiographic progression in patients with nr-axSpA.
Data from MEASURE 1 demonstrate sustained reduction of structural damage, whereas longitudinal data from cohort studies suggest long-term anti-TNF therapy is required before any positive effect on radiographic structural damage is observed. 60 Direct comparisons of structural progression results, however, cannot be made between these trials due to differences in study designs and populations. 60 Comparative data will become available from SURPASS: an ongoing, 2-year, randomized, active-controlled, head-to-head trial comparing prevention of structural damage progression with secukinumab (300–150 mg) versus an adalimumab biosimilar (40 mg) in biologic-naïve patients with r-axSpA. 61 Once completed, this trial will provide valuable information on the comparative prevention of radiographic progression by TNF inhibitors versus IL-17A inhibitors.
Objective signs of inflammation
Objective evidence of inflammation in patients with axSpA may be assessed non-invasively through observation of Berlin SIJ total oedema scores on MRI and elevated C-reactive protein (CRP) levels. 62 Elevated baseline CRP levels are associated with pain and fatigue symptoms and are a predictor of structural damage progression and response to TNF inhibitor therapy. 37 In MEASURE 2, the ratio of post-baseline level to baseline high-sensitivity CRP (hsCRP) level at week 16 was significantly lower with secukinumab 150 mg versus placebo (0.55 versus 1.13, p < 0.001); hsCRP levels remained low at 5 years.23,36 Similarly, in PREVENT, the mean change from baseline in hsCRP levels was lower for the secukinumab loading dose group than the placebo group in the overall population (0.64 versus 0.91, p = 0.0002). 33 Reductions in inflammation have also been observed with secukinumab through changes in Berlin SIJ total oedema scores on MRI.33,58 In MEASURE 1, the mean change from baseline in Berlin SIJ oedema score at week 16 was −1.30 for patients with r-axSpA receiving secukinumab 150 mg versus −0.17 for those receiving placebo. Responses were sustained through to 4 years.58,63 Meanwhile, in PREVENT, the change in SIJ edema score on MRI was significantly greater with secukinumab 150 mg loading dose versus placebo at week 16 (−1.68 versus −0.39, p < 0.001) and week 52 (−2.91) in the overall population. 33
Patient subgroup analyses in secukinumab RCTs
Patients with axSpA are a heterogenous population; therefore, subgroup analyses have been conducted to assess secukinumab’s efficacy across different patient groups. 64 Analyses of Asian populations have taken place across the MEASURE trials to determine whether secukinumab 150 mg is effective in Taiwanese (MEASURE 1), Japanese (MEASURE 2-J, an open-label phase III study) and Chinese (MEASURE 5) populations of patients with r-axSpA.32,65,66 In each patient population, secukinumab 150 mg provided sustained improvements in the signs and symptoms of r-axSpA.32,65,66
The burden of r-axSpA is reported to be higher in female patients than in male patients; many female patients show less improvement in axSpA outcome measures when treated with anti-TNF therapy versus male patients. 67 A post hoc analysis of data pooled from MEASURE 1–4 investigated differences in response to secukinumab according to sex. Similar efficacy outcomes were observed with secukinumab at weeks 16 and 52 in both female and male patients with r-axSpA (Table 2). 67
Summary of efficacy responses by sex in a post hoc analysis of pooled data from MEASURE 1–4. 67 .

ASAS, Assessment of SpondyloArthritis International Society; BASDAI, Bath Ankylosing Spondylitis Disease Activity Index; LD, loading dose; NL, non-loading dose.
Following the volume of clinical experience available, guidelines for axSpA treatment recommend TNF inhibitors first-line before IL-17A inhibitors.1,15 Therefore, the majority of patients receiving IL-17A inhibitors in practice may have previously had an inadequate response to anti-TNF therapy; it is important to understand treatment responses in these patients who have previously received other biologic therapy. In MEASURE 2, secukinumab 150 mg demonstrated sustained improvements in signs and symptoms of r-axSpA in anti-TNF-naïve and anti-TNF-IR patients (Figure 3). 36 In terms of nr-axSpA, as previously mentioned, the majority of patients (~90%) in PREVENT were anti-TNF-naïve. 33 Nevertheless, all secondary endpoints in the overall population, which included both anti-TNF-naïve and anti-TNF-IR patients, were met; this suggests that secukinumab 150 mg was effective in patients with nr-axSpA regardless of previous anti-TNF therapy. 33
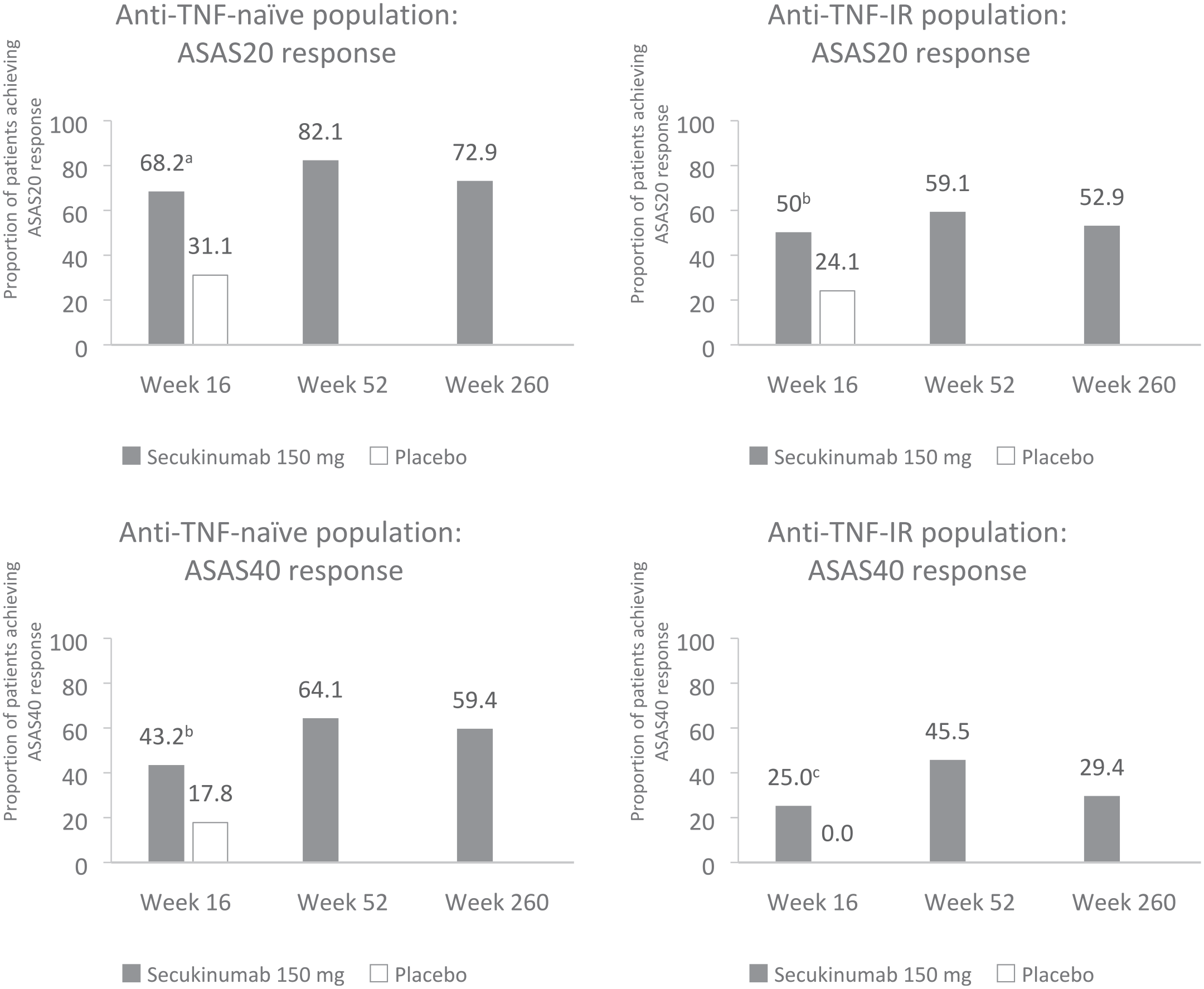
ASAS20/40 responses at weeks 16, 52 and 260 in MEASURE 2, stratified by anti-TNF status.36,68 Anti-TNF-naïve population: week 16, n = 44 (secukinumab 150 mg) and n = 45 (placebo); week 52, n = 39 (secukinumab 150 mg) and n = 42 (placebo); week 260: n = 37. Anti-TNF-IR population: week 16, n = 28 (secukinumab 150 mg) and n = 29 (placebo); week 52, n = 22 (secukinumab 150 mg) and n = 19 (placebo); week 260: n = 17. Missing data were imputed as NRI up to week 16. Observed data are presented at weeks 52 and 260.
AxSpA is strongly associated with the genetic marker HLA-B27; approximately 90–95% of Caucasian patients with r-axSpA express HLA-B27. 19 HLA-B27 status is a predictor of response to treatment in axSpA; hence, this is determined at baseline in axSpA clinical trials. 69 Pooled data from MEASURE 1–4 demonstrated that secukinumab was effective regardless of HLA-B27 status, although HLA-B27+ patients experienced increased therapeutic benefit compared with HLA-B27− patients. 69 In addition, higher age and longer disease duration are associated with a higher disease burden in axSpA. 70 In a pooled analysis of data from MEASURE 1–4, younger patients with shorter disease duration demonstrated greater responses in ASAS20/40, BASDAI, hsCRP and VAS disease activity/back pain scores with secukinumab than older patients with longer disease duration, emphasizing the importance of prompt diagnosis and treatment in axSpA. 70
As mentioned previously, elevated CRP levels are a predictor of treatment response in axSpA and are associated with pain and fatigue symptoms. In MEASURE 2, patients with r-axSpA were stratified according to their CRP status, classified as having normal (<5 mg/L) or elevated (⩾5 mg/L) levels of CRP at baseline. 37 This analysis demonstrated that secukinumab provided rapid and sustained relief of pain and fatigue symptoms, regardless of baseline CRP levels. 37
In PREVENT, patients with nr-axSpA were stratified by CRP and MRI status (positive or negative) at study entry to investigate how this affects the efficacy of secukinumab. Results demonstrated that secukinumab 150 mg provided numerically higher response rates versus placebo for ASAS40, BASDAI50, ASAS-PR and ASDAS-CRP ID in all CRP- and MRI-positive subgroups. 71 .
Safety
Biologic therapies used for managing axSpA have immunomodulatory potential and are intended for long-term use. Therefore, it is important to consider long-term safety of individual therapies when making axSpA treatment decisions. A total of 12,637 patients were included in an integrated clinical trial safety data set comprising secukinumab pooled data from 28 in patients with PsO, PsA and r-axSpA, with a cut-off date of 25 December 2018. 72 The r-axSpA cohort comprised 1140 patients from MEASURE 1–4 with a mean exposure of 1130.1 days. Adverse events (AEs) were reported as exposure-adjusted incident rates (EAIRs) per 100 patient-years. The safety profile from the integrated clinical trial safety data set was consistent with that reported in secukinumab RCTs (Table 3). 72 In PREVENT, secukinumab was well tolerated in patients with nr-axSpA. The safety profile was consistent with that of previously reported data in r-axSpA, with no new or unexpected safety signals and low EAIRs for serious infections (1.6), inflammatory bowel disease (0.9), malignancy (0.4) and uveitis (1.2) in secukinumab-treated patients with nr-axSpA (n = 369). 33
Selected AEs of interest with secukinumab from a 5-year pooled safety analysis. 72 .

AE, adverse event; CI, confidence interval; EAIR, exposure-adjusted incidence rate; IBD, inflammatory bowel disease; MedDRA, Medical Dictionary for Regulatory Activities; MACE, major adverse cardiovascular events; PsA, psoriatic arthritis; PsO, psoriasis; r-axSpA, radiographic axial spondyloarthritis; SD, standard deviation; SEC, secukinumab; URTI, upper respiratory tract infection.
‘Most common AEs’ defined as AEs that were reported to have the highest EAIR per 100 patient-years across the analysis.
Rates for system organ class.
Rates for high level term.
Rates for preferred term (PT; IBD for unspecified IBD).
Rates for Novartis MedDRA Query term.
Rates for standardized MedDRA query term – ‘malignancies and unspecified tumour’.
Although rates of inflammatory bowel disease (IBD) were low in MEASURE and PREVENT, there have been concerns regarding the association between IL-17A inhibition and new onset cases or exacerbations of IBD. 73 A large safety analysis including 794 patients with r-axSpA aimed to investigate the incidence of IBD cases throughout 4 years of treatment with secukinumab. Overall, IBD events were uncommon following secukinumab treatment in r-axSpA; new onset cases of IBD occurred in 1.1% of patients, and 0.5% of patients experienced exacerbations of existing IBD. 73 Nevertheless, secukinumab is not recommended for use in patients with IBD. If a patient develops signs and symptoms of IBD or experiences an exacerbation of pre-existing IBD, secukinumab should be discontinued and appropriate medical management should be initiated. 27
Immunomodulatory biologics for axSpA may associated with an increased risk of infections such as tuberculosis (TB). Anti-TNF biologics have linked to an increased risk of TB and latent TB infection (LBTI); meanwhile, there are limited data available that report the relationship between TB and biologics targeting other biologic pathways, such as IL-17. 74 A qualitative study comprising 12,319 patients with PsO (n = 8819), PsA (n = 2523) or r-axSpA (n = 977) showed that LBTI as an AE was uncommon after treatment with secukinumab. 74 Over 5 years, LBTI during secukinumab treatment was reported in 0.1% of patients (n = 13). 74
Overall, the data attained from these aforementioned RCTs affirm that secukinumab has a consistent safety profile in patients with axSpA.
Real-world evidence (RWE)
While RCT data are useful in treatment decision-making, strict inclusion and exclusion criteria mean that trial populations may not be truly representative of patient populations in clinical practice. 75 Therefore, evidence is needed to evaluate how RCT data translate into the real world. Because secukinumab gained approval in the EU and US (in 2016 for r-axSpA and in 2020 for nr-axSpA), multiple registries and studies have collected RWE for secukinumab.24,76 Here, we will focus on two key registries [EuroSpA and Consortium of Rheumatology Researchers of North America (CORRONA)] and studies (AQUILA and SERENA).77–80
EuroSpA is a collaboration between 16 European SpA registries. Data from EuroSpA have demonstrated that 81% of patients using secukinumab in clinical practice have previous experience with bDMARD, with a mean disease duration of 10 years. 81 We previously discussed that low disease activity is a treatment goal in axSpA; RWE from the EuroSpA registry has shown that low disease activity (defined as a BASDAI score <4) is maintained for at least 6 months in 49% of patients treated with secukinumab. 81 The CORRONA registry is a prospective, observational registry that includes patients with PsA and axSpA. 82 To date, CORRONA has published RWE for 106 patients with r-axSpA who have initiated biologic treatment, including 26 patients starting secukinumab. This registry has shown that secukinumab initiators had similar demographics, clinical outcomes, disease burden and patient-reported outcomes to those receiving other biologics. 82 No significant differences were observed for secukinumab initiators versus other biologic initiators in terms of mean scores for ASDAS (3.1 versus 3.2, p = 0.84), BASDAI (5.7 versus 5.4, p = 0.52) or BASFI (4.7 versus 4.2, p = 0.30). 82
AQUILA is an ongoing, non-interventional study based in Germany that aims to evaluate RWE for secukinumab treatment in PsA and r-axSpA. Data from this study found that secukinumab improves disease activity, physical functioning and QoL in patients with r-axSpA who were anti-TNF-naïve or anti-TNF-experienced with high adherence rates.80,83 Other findings from AQUILA include valuable insights into the real-world effect of secukinumab on functionality, as demonstrated by ASAS Health Index (ASAS-HI). For patients with r-axSpA receiving secukinumab, the mean ASAS-HI value decreased from 8.2 at baseline (n = 274, 88.1%) to 6.3 at week 52 (n = 156, 50.2%), indicating reduced disease burden. 84
SERENA is a Europe-based, ongoing, prospective, non-interventional study investigating the real-world, long-term use of secukinumab in more than 2900 patients with moderate-to-severe PsO, PsA or r-axSpA.77,85 An interim analysis of SERENA demonstrated that secukinumab is associated with high persistence rates in patients with r-axSpA, following high retention rates in the real-world setting after 2 years since enrolment in the study (n = 437, 78.9%) and after 2 years since initiation of treatment (n = 454, 84.8%). 85
Conclusions
AxSpA, comprising r-axSpA and nr-axSpA, covers a whole spectrum of disease with different courses. Therapy with bDMARDs should be considered for patients who are refractory to first-line NSAIDs. TNF and IL-17A inhibition were shown to be efficacious in patients whose disease is not adequately controlled by NSAIDs.
Data from MEASURE, PREVENT and SKIPPAIN have demonstrated that the IL-17A inhibitor secukinumab improves patient outcomes in line with the treatment goals defined by ASAS-EULAR and ACR-SAA-SPARTAN. Available RWE for secukinumab in axSpA will provide further insights into the real-world effectiveness of secukinumab. As yet, no head-to-head data are available for secukinumab versus other biologics in axSpA. Results of the ongoing trial SURPASS will provide valuable comparative data between secukinumab and anti-TNF therapy.
Supplemental Material
sj-docx-1-tab-10.1177_1759720X211041854 – Supplemental material for Secukinumab in axial spondyloarthritis: a narrative review of clinical evidence
Supplemental material, sj-docx-1-tab-10.1177_1759720X211041854 for Secukinumab in axial spondyloarthritis: a narrative review of clinical evidence by Jürgen Braun, Uta Kiltz, Björn Bühring and Xenofon Baraliakos in Therapeutic Advances in Musculoskeletal Disease
Footnotes
Acknowledgements
Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Adam Jowett of Ashfield MedComms, an Ashfield Health company, and funded by Novartis.
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship and/or publication of this article: JB received consulting fees from Novartis and Lilly, and speaker fees from Novartis, UCB Pharma, AbbVie and Gilead. UK declared no conflict of interest. BB received consulting fees from UCB Pharma and Gilead; speaker fees from UCB Pharma, Gilead, MSD, Biogen, Sanofi Genzyme and Amgen; support for attending meetings and/or travel from UCB, Gilead, Janssen and AbbVie; and participated in a data safety monitoring board or an advisory board for Gilead and Theramex. XB received speaker fees from AbbVie, Amgen, BMS, Chugai, Galapagos, Gilead, Lilly, MSD, Novartis, Pfizer, Roche, Sandoz and UCB Pharma.
Funding
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.