
Abstract
Synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome is a spectrum of heterogeneous diseases characterized by osteoarticular and dermatological manifestations. Osteitis and hyperostosis are core clinical manifestations in SAPHO syndrome, typically affecting multiple areas and possibly progressing to irreversible osteoarticular damage. Most patients with SAPHO have cutaneous involvement, mainly manifested as palmoplantar pustulosis and severe acne. Systemic manifestations are uncommon but occasionally reported. Epidemiological studies suggest the annual prevalence of SAPHO syndrome varies from 0.00144 in 100,000 in Japanese individuals to fewer than 1 in 10,000 in White individuals. The precise etiopathogenesis of SAPHO remains unclear, but it is generally considered an autoinflammatory syndrome that may be related to various etiologies, such as immune dysfunction, infection and genetic predisposition. Owing to the relapsing–remitting disease course, the goal of management is to improve clinical symptoms and prevent disease progression. Various treatments, including nonsteroidal anti-inflammatory drugs, conventional disease-modifying antirheumatic drugs, bisphosphonates, biologics, and antibiotics, are promising options for alleviating the disease.
Introduction
Synovitis, acne, pustulosis, hyperostosis, and osteitis (SAPHO) syndrome, first proposed by Chamot and colleagues 1 in 1987, is a spectrum of heterogeneous diseases characterized by osteoarticular and dermatological manifestations. The cardinal clinical manifestation in SAPHO syndrome is bone hyperostosis caused by sterile osteitis and osteomyelitis, with or without skin involvement. 2 Other clinical entities, such as sternocostoclavicular hyperostosis (SCCH), chronic recurrent multifocal osteomyelitis (CRMO), chronic nonbacterial osteomyelitis and mandibular sclerosing osteomyelitis, are also considered a part of SAPHO syndrome.3–5 Skin lesions of SAPHO syndrome showed neutrophilic infiltration, 2 which indicate potential links with autoimmune conditions, microbiome alterations, and genetic factors, as in other neutrophilic dermatoses. 6
In this review, we discuss recent advances concerning the epidemiology, etiopathogenesis, clinical manifestations, diagnosis, disease evaluation, and treatment of SAPHO syndrome.
Epidemiology
The precise epidemiological data for SAPHO syndrome remain unknown. The disease is distributed globally, with an estimated annual prevalence of fewer than 1 in 10,000 in White individuals and 0.00144 in 100,000 in the Japanese population. 2 To date, over 1000 cases of SAPHO syndrome have been reported worldwide,7,8 although the real prevalence is considered to be underestimated.
Etiopathogenesis
The precise etiopathogenesis of SAPHO remains unclear, but it is generally considered an autoinflammatory syndrome that may be related to various etiologies, such as immune dysfunction, 9 infection, 10 and genetic susceptibility. 11 An autoinflammatory origin was supported by the elevation of a series of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interleukin (IL)-1, IL-8, IL-17, and IL-18.12,13 The IL-23–helper T cell (Th)17 axis was also reported to be involved in SAPHO syndrome.14,15 Higher levels of receptor activator nuclear factor kappa-Β ligand (RANKL) were also reported in patients with active disease stage. 16 A recent study found a depletion of natural killer (NK) cells and an imbalance of Th17 and regulatory T (Treg) cells in patients with SAPHO, which may be related to immune inflammation. 17 The potential inflammation-mediated pathogenesis was further supported by the response to biologics targeting TNF-α, IL-1 and the IL-17–IL-23 axis. 18 In addition, several studies have isolated Propionibacterium acnes, Staphylococcus aureus, Haemophilus parainfluenzae, and actinomycetes in SAPHO osteitis lesions, 10 among which P. acnes was the most common species. 19 The interaction between transcription factor Forkhead Box O1 (FoxO1), P. acnes, NLRP3-inflammasome, and IL-1β may play a role in the development of osteitis. 13 The genetic susceptibility of SAPHO syndrome remains to be investigated. Family aggregation of SAPHO syndrome has been reported.20–24 Certain single nucleotide polymorphisms (MDM2 T309G, p53 G72C, rs6908425 T>C in CDKAL1) were found to be associated with SAPHO syndrome.25,26 Mutation in the NCSTN subunit of γ-secretase was identified in a patient with sporadic SAPHO with the manifestation of hidradenitis suppurativa. 27 Some genes on chromosomes 1 and 18 (LPIN2, PSTPIP2, and NOD2) were also found to be associated with conditions similar to SAPHO syndrome but not in SAPHO syndrome itself. 28 The connection between SAPHO syndrome and certain human leukocyte antigens (HLAs; including HLA-A26, HLA-B27, HLA-B39, and HLA-B61) has been controversial.2,29–33 The potential etiologies of SAPHO syndrome are summarized in Table 1.
Potential etiologies of SAPHO syndrome.

FoxO1, Forkhead Box O1; HLA, human leukocyte antigen; IL, interleukin; NK, natural killer; RANKL, receptor activator nuclear factor kappa-B ligand; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis; Th, helper T cell; TNF, tumor necrosis factor; Treg, regulatory T cell.
Clinical manifestations
Osteitis and hyperostosis are the core clinical manifestations in SAPHO syndrome, which typically affect multiple areas and may progress into irreversible bone and joint damage. 34 The most commonly affected area is the anterior chest wall (ACW), followed by the axial skeleton (including the spine and the sacroiliac joints), the long bones of the extremities, the irregular bones (such as mandible), and the peripheral joints. ACW involvement, which occurs in 65–90% of patients,35–38 is highly characteristic of SAPHO syndrome. More specifically, the typical affected structures of the ACW include the sternocostal joints, the sternoclavicular joints and the costoclavicular ligament. The surrounding soft tissue can become reddish and swollen, leading to the compression of nearby structures. 37 Approximately 32–52% of patients have axial involvement, which manifests as pain in the spine or gluteal region.36,37 This group of patients usually presents with more serious clinical manifestations and requires more aggressive treatment. 8 Peripheral bone and joint involvement is common in patients with SAPHO, which occur in 65.8–82.9% of patients.8,9
Most patients with SAPHO have cutaneous involvement, mainly manifested as palmoplantar pustulosis (PPP) and severe acne (SA), which are shown in Figure 1. 34 PPP, which is recognized as a special type of psoriasis, is characterized by chronic, recurrent, sterile, small pustules and vesicles. 39 Among the 354 Chinese patients in the cohort, 94.6% reported skin involvement, of whom 91.9% had PPP, 14.3% had SA, and 15.8% had psoriasis vulgaris. 8 Psoriatic nail disease has also been reported in some patients. 9 Skin manifestations can occur at any stage of the disease or be absent. A majority of the patients (over 70%) develop both cutaneous and osteoarticular symptoms within 2 years,8,40,41 although longer intervals have been reported.40,42
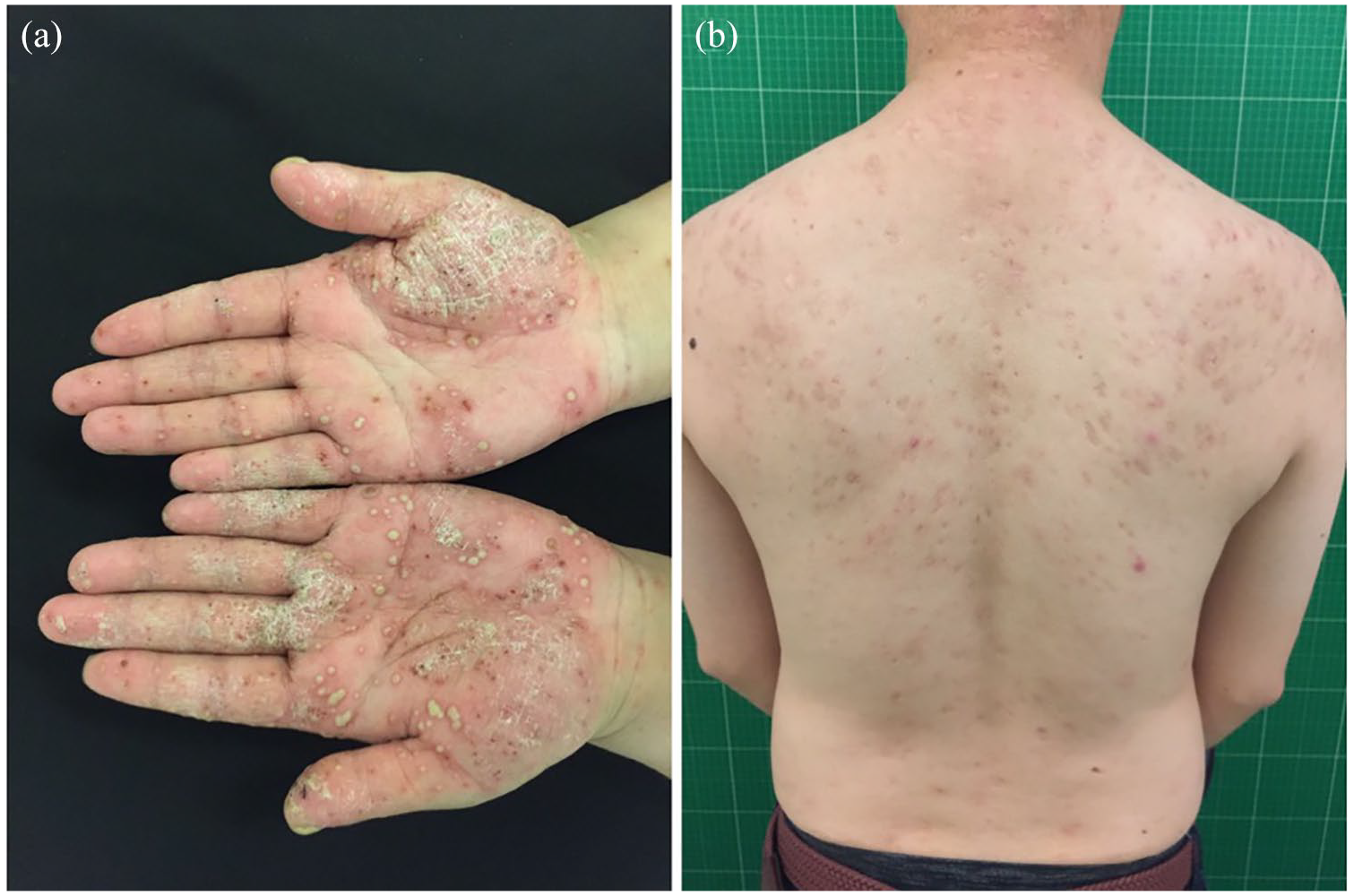
Characteristic cutaneous manifestations of SAPHO syndrome: (a) palmoplantar pustulosis (PPP); (b) severe acne (SA).
Systemic manifestations such as fever and elevation of inflammatory markers are uncommon but occasionally reported. 7 Other extra-articular manifestations include inflammatory bowel disease, 43 pulmonary involvement, 44 venous thrombosis (most commonly affecting the subclavian vein), 45 dura mater hypertrophy, 46 and uveitis. 47
Diagnosis
The most widely applied diagnostic criteria were proposed by Benhamou and colleagues 48 in 1988, who established the diagnosis of SAPHO syndrome on the basis of clinical manifestations and radiological examinations, including bone, articular or skin manifestations. Another commonly used diagnostic criterion was proposed in 1994 and revised in 2003 by Kahn and Kahn, 49 who established the diagnosis of SAPHO syndrome mainly on the basis of clinical symptoms. The diagnostic criteria are summarized in Table 2.
Diagnostic criteria for SAPHO syndrome.

ACW, anterior chest wall; CRMO, chronic recurrent multifocal osteomyelitis; PPP, palmoplantar pustulosis; SA, severe acne; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis.
Currently, no specific clinical features or laboratory findings have been verified to confirm the diagnosis of SAPHO syndrome. Based mostly on clinical and radiological manifestations, the diagnosis of SAPHO syndrome is, to some extent, a diagnosis of exclusion. However, for patients without the typical pattern of skin lesions and osteoarticular involvement (e.g. with long/flat bone involvement), the diagnosis remains challenging.
Differential diagnosis
Considering the complexity of manifestations of SAPHO syndrome, other inflammatory, infectious, and neoplastic etiologies should be considered. For example, Sonozaki syndrome [or pustulotic arthro-osteitis (PAO)], which belongs to the group of psoriatic arthritis, includes PPP as well as arthro-osteitis, which typically involves the sternoclavicular joint.51,52 Considering the overlaps with other clinical entities, patients with SAPHO syndrome should be distinguished from patients with other osteoarticular diseases, with or without skin manifestations, and other systematic autoinflammatory diseases (Table 3).
Differential diagnosis of SAPHO syndrome.
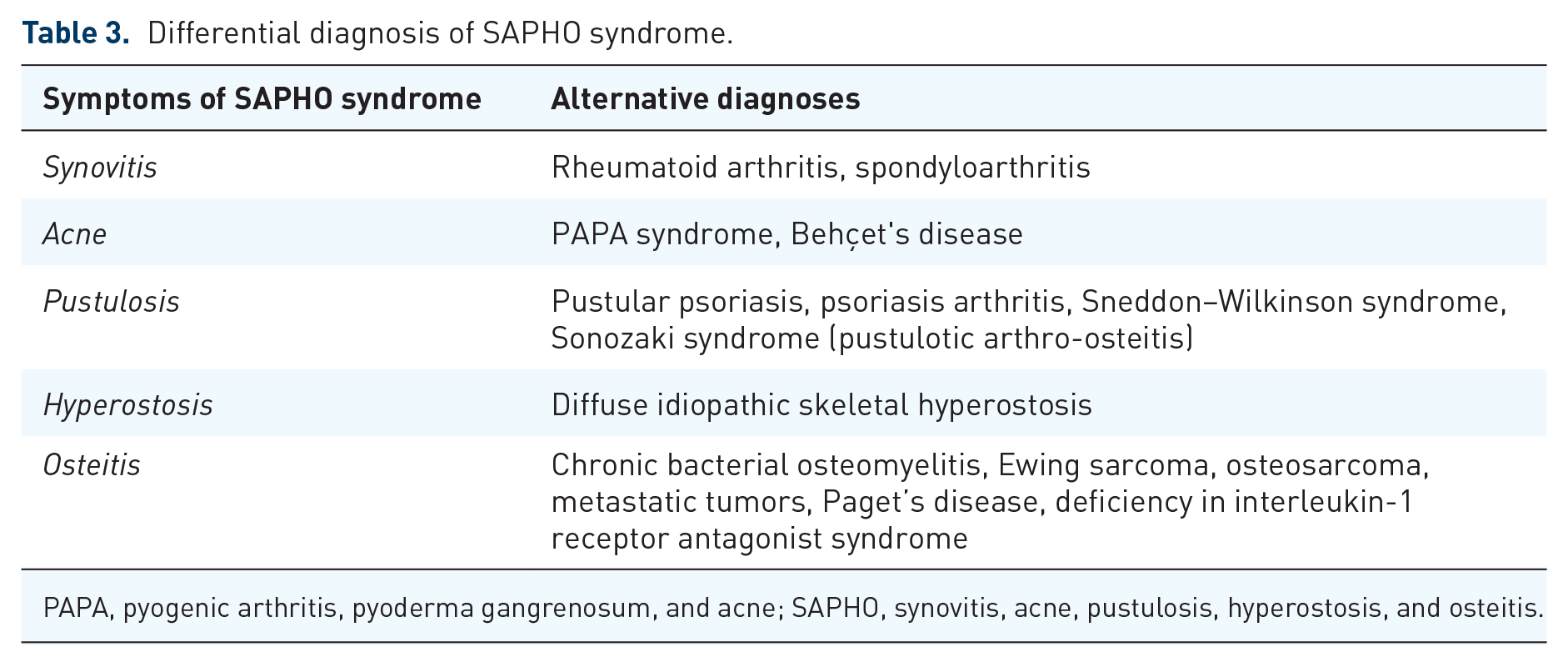
PAPA, pyogenic arthritis, pyoderma gangrenosum, and acne; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis.
Disease evaluation
A comprehensive evaluation is important for the diagnosis and management of SAPHO syndrome. As shown in Figure 2 and Table 4, the disease shows unique patterns of vertebral involvement by multiple imaging techniques, 53 yet manifestations may vary with the age of onset, the affected regions, and the course of disease. A series of inflammatory markers and bone metabolites have been found to be useful in quantifying the inflammatory status of SAPHO syndrome. In addition, patient-reported outcomes (PROs) have been widely used in the evaluation of a variety of chronic diseases, including SAPHO syndrome, which better reflect the functional impairment of the disease.

Characteristic radiological manifestations of SAPHO syndrome. CT revealed bone cortical destruction and osteosclerosis of bilateral sternoclavicular joints, and swelling of the surrounding soft tissues (a). Whole spinal CT showed bone destruction in multiple vertebrae (b). MRI demonstrated multiple patchy, short T1 and long T2 (d) signals of lumbar vertebrae. WBBS showed increased radioactivity in the left sternoclavicular joint, the left first anterior rib, the second and fourth lumbar vertebrae, and the right iliac joint (e). PET/CT showed bone destruction and increased glucose metabolism in left clavicle bone (f) and vertebra (g).
Imaging techniques used in SAPHO syndrome.

ACW, anterior chest wall; CT, computed tomography; MRI, magnetic resonance imaging; PET, positron emission tomography; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis; WBBS, whole-body bone scintigraphy.
Imaging assessment
X-ray and computed tomography
Conventional X-ray and computed tomography (CT) scans are classical choices to assess bone hypertrophy and osteitis. X-ray imaging can detect osteoarticular changes in the long bones at relatively late stages, with characteristic manifestations including irregular bone morphology, cortical thickening, increased density of the medullary cavity with or without low-density destruction areas.36,37 However, the low sensitivity of X-ray to detect lesions at early stages, especially those in the ACW, may lead to delayed diagnosis. 54
The high density and spatial resolution of a CT scan facilitates its use in detecting osteoarticular lesions that are difficult to observe on X-ray. The ability to clearly reveal bone hyperplasia at the attachment point of the costoclavicular ligament at an early stage makes CT an important diagnostic tool for SAPHO syndrome. The whole-spine CT can clearly demonstrate the distinct characteristics of spinal lesions, in which the location of vertebral corner and ‘kissing’ involvement pattern are indicative of SAPHO syndrome. 55
Magnetic resonance imaging
With an advantage over CT in assessing early and active disease, magnetic resonance imaging (MRI) can be employed as a guide for treatment and follow up. Bone marrow edema shown on MRI suggests an active disease state, presenting as a low signal on T1WI, a high signal on T2WI and short-TI inversion recovery images, and marked enhancement on enhanced scans. 37
Whole-body bone scintigraphy
Whole-body bone scintigraphy (WBBS) has the advantages of demonstrating multifocal osteoarticular lesions at the same time and finding clinically insidious lesions. 57 The typical ‘bull’s head’ pattern on WBBS, that is, the high uptake of the ‘sternocostoclavicular’ joint and the sternal angle is highly specific for SAPHO syndrome. 58 The bull’s head sign is especially important for the diagnosis of patients with nontypical SAPHO who have atypical cutaneous lesions or lack cutaneous manifestations. 59 Based on WBBS manifestations, the osteoarticular involvement in SAPHO syndrome can be categorized into three distinct patterns, that is, the spinal type, the costal type and the sternoclavicular type. 64 However, both active and chronic lesions manifest as high uptake areas on WBBS, which means WBBS cannot determine the disease activity of lesions.
Positron emission tomography/CT
Positron emission tomography (PET)/CT can demonstrate the location and distribution of inflammation in osteoarticular lesions. 60 The typical PET/CT findings of SAPHO syndrome are multiple skeletal lesions in the ACW or spine with low to moderate 18F-FDG uptake and concurrent osteolysis and osteosclerosis. 61 In addition, PET/CT serves as an important diagnostic tool to differentiate benign lesions from bone metastasis.62,63 However, the ability of PET/CT to determine disease activity in SAPHO syndrome requires further investigation.
Laboratory tests
Elevation of nonspecific inflammatory markers [e.g. erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)] can be seen in the active phase of the disease.8,65 During the improvement and relapse periods, serum amyloid A appeared to show higher sensitivity than CRP. 66 An anomaly of proinflammatory and anti-inflammatory cytokine expression was observed in active SAPHO patients, presenting as higher levels of serum TNF-α, IL-6, IL-8, and IL-17A levels.12,16 Raised immunoglobulin (Ig)G4 levels are reported in 23% of patients with SAPHO and are associated with higher disease activity. 67 Abnormal levels of bone metabolites may also occur, manifesting as an increase in the osteoclast marker β-isolated C-terminal peptide (β-CTX) and a decrease in the osteoblast marker osteocalcin. 68 An increased RANKL level and RANKL/osteoprotegerin (OPG) ratio are seen in patients with active SAPHO. 16 The frequency of HLA-B27 was not significantly higher than that in the normal population. 9 Potential laboratory markers of SAPHO syndrome are summarized in Table 5.
Potential laboratory markers of SAPHO syndrome.

CRP, C-reactive protein; ESR, erythrocyte sedimentation rate; IL, interleukin; OPG, osteoprotegerin; RANKL, receptor activator nuclear factor kappa-Β ligand; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis; TNF, tumor necrosis factor.
Patient-reported outcomes
Studies based on PROs on SAPHO syndrome are limited. A nationwide patient survey in Germany showed a high impact of SAPHO syndrome on patients’ general health and quality of life, mainly due to musculoskeletal symptoms. 69 Further investigation is needed to explore the usefulness of PROs to reflect the treatment efficacy of drugs and the functional status of patients.
Treatment
Strategy
To date, most studies on the treatment of SAPHO syndrome are case reports, case series, or observational cohorts. Evidence based on randomized clinical trials is still lacking. As a result, no consensus has been reached on the treatment of SAPHO syndrome.
The first and primary goal of treatment is the improvement of clinical symptoms, including ostealgia and skin rash. Second, treatment should take effect in slowing the progression of joint involvement and the regression of articular function, thereby improving patients’ quality of life in the long term. Table 6 summarizes the current treatment options on SAPHO syndrome.
Treatment on SAPHO syndrome.

+, general improvement without specifying certain symptoms; –, no improvement; ±, contradictory results reported by previous studies.
DMARD, disease-modifying antirheumatic drug; IL, interleukin; MRI, magnetic resonance imaging; N/A, not available/applicable; NSAID, nonsteroidal anti-inflammatory drug; PDE-4, phosphodiesterase type 4; PPP, palmoplantar pustulosis; SAPHO, synovitis, acne, pustulosis, hyperostosis, and osteitis; TNF, tumor necrosis factor.
Nonsteroidal anti-inflammatory drugs
Nonsteroidal anti-inflammatory drugs (NSAIDs) are generally regarded as first-line medications for pain relief and symptom control of SAPHO syndrome. NSAIDs work quickly and remarkably for patients at the diagnostic stage of disease. However, NSAID monotherapy has shown limited effects in patients with extensive osteomyelitis. 92 The gastrointestinal side effects of NSAIDs should be taken into consideration.
Disease-modifying antirheumatic drugs
Conventional disease-modifying antirheumatic drugs (cDMARDs) are usually recommended as second-line options, and the responses to cDMARDs vary among different patients. For patients with peripheral joint involvement and relatively low levels of axial spine joint involvement, the administration of methotrexate (MTX) could be effective.72–74 Improvement of peripheral osteoarticular symptoms and pleural effusions could occur within several weeks to months. However, the efficacy of MTX for the treatment of osteitis, osteomyelitis or enthesitis is uncertain. Other cDMARDs, including sulfasalazine, hydroxychloroquine, leflunomide, thalidomide, and colchicine, have been reported to be beneficial in SAPHO syndrome based on case reports or case series.7,40,93 Further evidence is needed to determine the efficacy of these agents.
Corticosteroids
Systematic or intra-articular corticosteroids work quickly but transiently. 94 Relapses tend to occur when treating cutaneous lesions and appear to be even more serious than before. 95 Taking adverse effects into consideration, corticosteroids are preferably used as a bridge treatment in moderate doses and for a short term.
Bisphosphonates
Bisphosphonates consistently inhibit osteoclast activity and exert anti-inflammatory effects. 96 The use of intravenous bisphosphonates (especially pamidronate) for SAPHO syndrome has been reported, demonstrating partial or full remission of both ostealgia and cutaneous lesions.29,78
Targeted drugs
Use of anti-TNF-α biologics has been frequently reported. Previous reports of the treatment of SAPHO syndrome with infliximab, 79 adalimumab,75,80 and etanercept 97 have proved the effectiveness of these treatments for osteoarticular and cutaneous lesions. However, during anti-TNF-α therapy, some patients develop new paradoxical skin lesions, which present as psoriasiform scaly plaques or pustular lesions.98,99
Other less commonly used biologics, such as anakinra, 89 ustekinumab, 15 secukinumab,15,85 and apremilast, 91 have also shown beneficial effects in some patients. Although administration of tocilizumab, an anti-IL-6 agent, showed efficacy to some extent, aggravation or development of lesions were quite common. Existing observations indicate that tocilizumab may not be an ideal option for SAPHO syndrome, and it should be considered with caution.86–88
Tofacitinib, a small-molecule nonspecific JAK 1 and JAK 3 inhibitor, was used to treat a patient refractory to SAPHO and demonstrated amelioration in terms of clinical symptoms, inflammatory parameters, and MRI. 90
Antibiotics
Since infection by P. acnes is thought to be a possible pathogenetic trigger of SAPHO syndrome, especially for patients with SA, 13 antibiotics have also been considered for treatment. Tetracyclines, 29 clindamycin, 100 and azithromycin 29 have been reported to be successful in treating some cases of acne but show little efficacy against other symptoms. Skin involvement in SAPHO syndrome mainly manifests as PPP, in which condition antibiotics show an effect that is curative but not as dramatic as that in the treatment of acne.
Prognosis
SAPHO syndrome is a chronic disease that typically follows a relapsing–remitting disease course. 101 Some patients undergo one or two attacks with eventual spontaneous resolution. Others experience a prolonged and sometimes disabling evolution with the appearance of new cutaneous or osteoarticular manifestations. The prognosis of SAPHO syndrome is generally good.40,102 However, for patients with pathological fractures in the vertebral bodies or clavicle, the prognosis is not optimistic. 103
Conclusion
SAPHO syndrome is a chronic inflammatory disorder characterized by osteoarticular and dermatological involvement. Osteitis and hyperostosis are considered the core clinical manifestations, which mainly affect the axial skeleton with characteristic involvement of the ACW. A wide spectrum of neutrophilic dermatoses is associated with the disease, with PPP and SA most commonly observed. The pathogenesis remains unclear, yet SAPHO syndrome is considered an autoinflammatory disorder related to a variety of genetic and environmental factors and immune dysregulation. Diagnosis is still challenging due to the clinical heterogeneity of the disease. A comprehensive evaluation including imaging and laboratory examinations is important for early diagnosis and treatment. A variety of therapeutic options, including bisphosphonates and targeted drugs, have been proposed to alleviate symptoms and prevent disease progression.
Footnotes
Acknowledgements
Shuang Liu and Mingwei Tang contributed equally to this manuscript.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the CAMS Initiative for Innovative Medicine (grant number 2017-I2M-3-001), the Capital Medical Research and Development Fund (grant number 2016-4-40112), and the National Key Research and Development Program of China (grant number 2016YFC0901500).
Conflict of interest statement
The authors declare that there is no conflict of interest.
Clinical trial registration
Not applicable.