
Abstract
Background:
Histopathological evaluation, TNM classification, and mediastinal staging via endobronchial ultrasound-guided transbronchial needle aspiration provide essential guidance for therapeutic decision-making in non-small cell lung cancer (NSCLC). Genomic profiles of primary tumors (PT) and matched mediastinal lymph nodes (MLN) can uncover occult metastases, refine recurrence risk assessment, and support personalized treatment strategies in lung adenocarcinoma (LUAD).
Objectives:
The study aimed to investigate clinically relevant somatic variants in formalin-fixed, paraffin-embedded (FFPE) PT and scrapings of MLN cytological slide samples using next-generation sequencing (NGS) and correlated the findings with overall survival (OS).
Design:
We retrospectively analyzed the genomic profile of PT and MLN from Brazilian LUAD patients between 2013 and 2020.
Methods:
Genomic DNA (gDNA) was extracted from PT and MLN matched samples of 32 patients (n = 64). Targeted NGS was performed using the SureSelect XTHS2 panel. We analyzed OS using Kaplan–Meier curves, and risk factors for mortality using univariate and multivariate Cox regression models.
Results:
Clinically significant or potentially significant variants were identified in 72% of PT and 75% of MLN, totaling 46 and 51 variants, respectively. The most frequent variants in PT involved EGFR (39.3%), TP53 (28.6%), ATM, and KRAS (21.4% in both), whereas in MLN, EGFR (35.7%), TP53 (32.1%), KRAS and BRAF (14.3% in both), and ATM (10.7%) have predominated. Co-mutations were detected in 31.3% of PT and 43.8% of MLN, with variant discordance between PT-MLN in 82.2% of cases.
Conclusion:
Cox regression analysis demonstrated that the presence of co-mutations in MLN was a co-factor associated with poorer OS, while no significant associations were observed for PT variants. These findings highlight distinct genomic profiles between PT and MLN. Integrating the molecular profile of MLN into staging may improve prognostic accuracy and guide treatment decisions in LUAD patients.
Plain language summary
Various tests are used to diagnose and treat non-small cell lung cancer, including histopathology and the classification of tumors based on their size, location, and the presence of distant metastases and lymph node involvement. Ultrasound-guided biopsies are also employed. These tools assist in planning the best treatment for patients. Additionally, analyzing DNA from tumors removed during surgery and from lymph nodes obtained via endobronchial ultrasound can uncover hidden small metastases, help assess the risk of cancer recurrence, and guide personalized therapies. In this study, we examined DNA from paired samples of primary tumors and lymph nodes from 32 patients with lung adenocarcinoma (a total of 64 samples). We utilized advanced sequencing techniques to identify genetic alterations relevant to lung cancer treatment. We found that many of these alterations were present in a large proportion of the samples, especially in genes such as EGFR, TP53, ATM, and KRAS. Our results showed that the combination of these genetic alterations within the same patient was linked to lower survival rates. Specifically, mutations in TP53 and ATM, as well as their combinations in lymph nodes, were associated with a poorer prognosis. Furthermore, we observed that the genetic profiles of primary tumors and lymph nodes differed in most cases. Our study demonstrates that analyzing the genetics of lymph nodes can help predict cancer recurrence, detect small metastases, and determine the most effective personalized treatment for each patient.
Introduction
Lung cancer (LC) remains the leading cause of cancer incidence and mortality worldwide. In Brazil, LC is the fourth most common malignancy, with a 5-year overall survival (OS) of approximately 15%–21%, closely aligning with global statistics.1,2 Non-small cell lung cancer (NSCLC) represents approximately 85% of LC cases and is characterized by substantial genomic heterogeneity. 3 Among it is subtypes, lung adenocarcinoma (LUAD) is the most prevalent and presents metastatic disease in nearly 50% of cases at diagnosis and an estimated 5-year survival rate of only 15%.4–6 The occurrence of metastasis remains the primary cause of cancer-related mortality, 7 and concerning NSCLC, mediastinal lymph node (MLN) involvement plays a critical role in staging, prognosis, and therapeutic decision-making, as nodal metastasis often precludes surgical intervention and is associated with higher recurrence and reduced survival rates. 8
Endobronchial ultrasound-guided transbronchial needle aspiration (EBUS-TBNA), a minimally invasive technique introduced in the 1990s, is currently recommended by international guidelines for mediastinal staging.9–11 When combined with imaging modalities such as PET-CT, EBUS-TBNA enables accurate sampling of lymph nodes, even those smaller than 1 cm, using high-frequency ultrasound and real-time visualization. Rapid on-site evaluation (ROSE) may be performed concurrently to ensure sample adequacy. 12 Studies have demonstrated that EBUS-TBNA offers sensitivity, specificity, and diagnostic accuracy comparable to mediastinoscopy (81%, 100%, and 93%, respectively).12–14
At the molecular level, LUAD is frequently associated with somatic alterations in key oncogenes and tumor suppressor genes, including KRAS, TP53, ATM, BRAF, and EGFR. KRAS mutations are observed in up to 30% of LUAD cases, particularly among smokers. While historically considered undruggable, recent advances in targeted therapies such as Sotorasib and Adagrasib have shown efficacy against specific mutations like KRAS G12C.15–18
The molecular profile of PT was considered the standard for evaluating somatic variants in cancer patients; however, emerging evidence indicates that MLN may harbor clinically significant variants not detected in the primary tumor (PT), reflecting intratumoral heterogeneity and clonal evolution. The discordance between PT and MLN genomic profiles has important clinical implications: actionable mutations present in MLN could influence variants in the TP53 gene, a crucial tumor suppressor, which are also common in LUAD and have been linked to poor prognosis. Additionally, alterations in ATM, a DNA damage response kinase, may contribute to genomic instability and disease progression, with potential implications for therapeutic resistance.16,17
Clinically significant BRAF mutations, including V600E, are present in approximately 3%–8% of LUAD cases and are more common among current or former smokers. Targeted therapies using tyrosine kinase inhibitors (TKIs) have shown promise in patients harboring BRAF alterations.19,20 Amplifications and activating mutations in the EGFR gene are more frequently observed in non-smoking women of Asian descent but are also clinically relevant in diverse populations. These mutations are associated with tumor progression and sensitivity to EGFR-targeted TKIs.21,22
Therefore, identifying these genomic alterations is critical in improving diagnostic precision, predicting prognosis, and guiding personalized treatment. Moreover, integrating molecular profiling with TNM staging may enhance clinical decision-making and patient outcomes.23,24 Thus, this study aimed to evaluate genomic variants in PT and MLN samples from a cohort of Brazilian patients with LUAD using next-generation sequencing (NGS). We then compared the genomic profiles and assessed the impact of these variants on patient survival.
Materials and methods
Study design and patients
This retrospective study included 32 patients diagnosed with stage I–III LUAD according to the 7th edition of the TNM classification. 25 Eligible patients had not received any prior chemotherapy, showed no evidence of metastasis or recurrence at diagnosis, and had paired samples available of their PTs and MLN aspirates, which were collected via EBUS-TBNA. The samples were obtained from the Instituto do Cancer do Estado de Sao Paulo—Hospital das Clinicas da Universidade de Sao Paulo between 2013 and 2020, with a minimum cellularity threshold of ⩾20% required.
Patients were divided into two groups: Group A included patients with histologically negative MLN (MLN−), while Group B included patients with histologically positive MLN (MLN+).
An experienced pulmonary pathologist reviewed all the histologic slides. This study included formalin-fixed, paraffin-embedded (FFPE) tissue sections that were stained with hematoxylin and eosin as well as Papanicolaou-stained cytology slides evaluated by ROSE. This study was approved by the Institutional Ethics Committee (approval number: 3.004.983/2018) and was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines. Clinical data were obtained from the Brazilian Lung Cancer Registry. In addition, the reporting of this study is according to the STROBE statement (Supplemental Appendix 1).
Sample processing, extraction, quantification, and integrity assessment of genomic DNA
The processing of FFPE samples, adaptation to the genomic DNA (gDNA) purification protocol, processing of histological and cytological slides, and quantification and assessment of DNA integrity were performed in the manner previously suggested by Faria et al. 26
Library preparation, sequencing, and variant analysis
NGS libraries were prepared using the SureSelect® XTHS2 Custom Panel (Agilent Technologies, Santa Clara, CA, USA), which targets 107 genes involved in NSCLC and metastasis-related pathways. We optimized the NGS protocol with increased cycling to introduce samples with a DNA Integrity Index (DIN) ⩽3 for better gDNA amplification. After the final assessment of gDNA integrity by automated electrophoresis, only samples with a DIN ⩾3 proceeded to library assembly for sequencing.
Sequencing was conducted at the Large-Scale Sequencing Laboratory of the University of Sao Paulo Medical School (SELA-FMUSP). Only samples with DIN ⩾3 were included. Libraries were normalized to 1.8 pM, denatured with 0.1 N NaOH, and sequenced using the NextSeq 500/550 v. 2.5 high-output kit (2 × 150 bp paired-end reads) on the Illumina NextSeq 500 platform (Illumina Inc., San Diego, CA, USA). The proportion of sequence reads carrying a specific variant (Variant Allele Frequency, VAF) considered in the study was ⩾0.05. A barcode index (double index, paired end) was used as the unique molecular identifiers to suppress duplication errors and amplification artifacts. The reads were aligned to the human genome reference (GRCh37/hg19). Variants with ⩾10 sequence reads (read depth) were included in the study. BAM files were uploaded to Illumina’s BaseSpace Sequence Hub, with a minimum coverage of 1000×, to detect allele frequencies <1%. FastQ files were processed on the Varstation 3.0® online platform (VarsOmics, Albert Einstein Hospital, São Paulo, Brazil). We used a somatic pipeline with strand bias filters such as GATAK (UnifiedGenotyper and HaplotypeCaller), SAMtools, VarScan, and smCounter. In the next step, variant calling and annotation, and reports were generated for analysis. The variants were initially analyzed and validated according to the ACMG guidelines, 27 and interpreted and defined according to the 2017 AMP/ASCO/CAP guidelines, 28 as follows:
Tier I: strong clinical significance (diagnostic, prognostic, therapeutic)
Tier II: potential clinical significance
Tier III: variants of uncertain significance
Tier IV: benign or likely benign.
Only Tier I and II variants were included in this study. Additional curation for somatic variants was performed with the assistance of online databases OncoKB, 29 COSMIC, 30 VarSome, 31 and Franklin by Genoox platform, 32 with filters set for “lung cancer,” “lung adenocarcinoma,” and “general cancer.”
Among the 88 initial samples (44 patients), only 64 met the quality criteria for sequencing (21 from Group A, 11 from Group B). Sequencing revealed a 1000× mean depth with a Q30 quality score of 92.5%. Four Group A patients exhibited no Tier I/II alterations classified by AMP/ASCO/CAP guidelines 28 but remained in the clinical dataset.
Statistical analysis
Clinical data were retrieved from the Tasy and REDCap databases of the Brazilian Lung Cancer Registry. Associations between clinical characteristics and variant presence (pathogenic or likely pathogenic) in PT and/or MLN samples were assessed via the Chi-square test or Fisher’s exact test, as appropriate. Survival differences were evaluated using the log-rank test with Kaplan–Meier survival curves. The OS was calculated in terms of the time period between the date of diagnosis, and the date of death from any cause, for all patients.
Survival analysis was performed using the Kaplan–Meier method. The association between the OS rate and other covariates was evaluated using the Cox proportional hazards model. To construct the multivariate model, we considered any clinically relevant parameter or those significant in the univariate analysis.
All the statistical analyses were performed via IBM SPSS Statistics v. 25.0 (IBM Corp., Armonk, NY, USA) and Excel, Office Professional Plus 2019 (Microsoft Corporation, Redmond, WA, EUA). A p-value < 0.05 was considered to indicate statistical significance.
Results
Patient and sample characteristics
In this study, we performed NGS on paired PT and MLN samples from 32 Brazilian patients diagnosed with LUAD. The group of patients studied presented an equal distribution of male and female patients (50% each; n = 16), with a median age of 65 years (range: 47–80 years). Most patients reported a history of tobacco use, and only six individuals (18.8%) identified as individuals who had never smoked.
Histologically, the most prevalent LUAD subtype was acinar (40.6%, n = 13), followed by the solid (31.3%, n = 10), lepidic (9.4%, n = 3), mucinous (3.1%, n = 1), and papillary (3.1%, n = 1) subtypes. In four patients (12.5%), the tumors were classified as not otherwise specified due to limited sample material, precluding a more specific classification.
According to the 7th edition of the TNM staging system, 25 59.4% of patients (n = 19) were diagnosed with stage IIIA disease, and 34.4% (n = 11) presented with histologically confirmed lymph node metastasis. The mean tumor diameter was 3.6 cm (range: 1.5–10.4 cm).
During the follow-up period (median: 40.9 months, range: 1.6–82.4 months), 59.4% of patients (n = 19) developed distant metastasis. Disease recurrence was observed in 56.3% (n = 18) of patients, and 50% of the patients (n = 16) died.
In terms of treatment modalities, surgical resection was performed in 68.7% (n = 22) of patients, chemotherapy was indicated in 71.9% (n = 23) of patients, and radiotherapy was administered to 21.9% (n = 7) of patients. Among the 23 patients who received chemotherapy treatment, 91.3% (n = 20) received platinum-based treatment combined with other drugs (paclitaxel, docetaxel, vinorelbine, gemcitabine), 8.7% (n = 2) received treatment with gefitinib single agent.
Among the PT samples analyzed in the study, 10 were non-surgical: 7 samples were obtained from transthoracic biopsies and 3 from histological slide scrapings, which were used to extract genomic material. Of the 22 surgical cases, FFPE material from tumor resection was used in 16 cases, and histological slide scrapings were used in 6 cases. The 32 MLN samples were obtained by EBUS from cytological slide scrapings. All samples were treated with uracil-DNA glycosylase to eliminate uracil residues produced by cytosine deamination caused by FFPE processing. Only samples with tumor cellularity ⩾20% were included in the study.
The MLN samples were histologically classified as MLN+ in 11 patients and MLN– in 21 patients.
A summary of the clinical and pathological characteristics of the patients is provided in Table 1.
Demographic and clinicopathological characteristics of patients diagnosed with LUAD (n = 32).

Some cases lacked follow-up information: smoke status (1), smoking load (7), tumor size (2).
7th International Association for the Study of Lung Cancer (Ref. 25).
LUAD, lung adenocarcinoma; NOS, not otherwise specified.
Genomic variant profile
NGS analysis of somatic variants with strong or potential clinical significance (TIERs I and II) revealed a diverse spectrum of genomic alterations across the casuistic, and were demonstrated in Supplemental Table and Figure 1. The tables show the characterization of the genomic profile in Group A MLN− (Supplemental Table 1) and Group B MLN+ (Supplemental Table 2), organized by patient. They include information on genomic loci, Single-Nucleotide Polymorphism database identifiers, altered nucleotides and proteins, variant types, VAFs, read depths, and classifications according to the AMP/ASCO/CAP guidelines for somatic cancer variants. 28 In Group A, 41.1% of the detected variants were classified as TIER I, while 57.9% were categorized as TIER II. Among the MLN samples from the same group, TIER I variants were identified in 57.15% of cases, with the remaining 42.85% classified as TIER II. In Group B, 55.22% of variants in MLN samples were classified as TIER I, and 44.8% as TIER II. In the corresponding PT samples from this group, TIER I variants were observed in 61.5% of cases, whereas TIER II variants accounted for 38.5%.
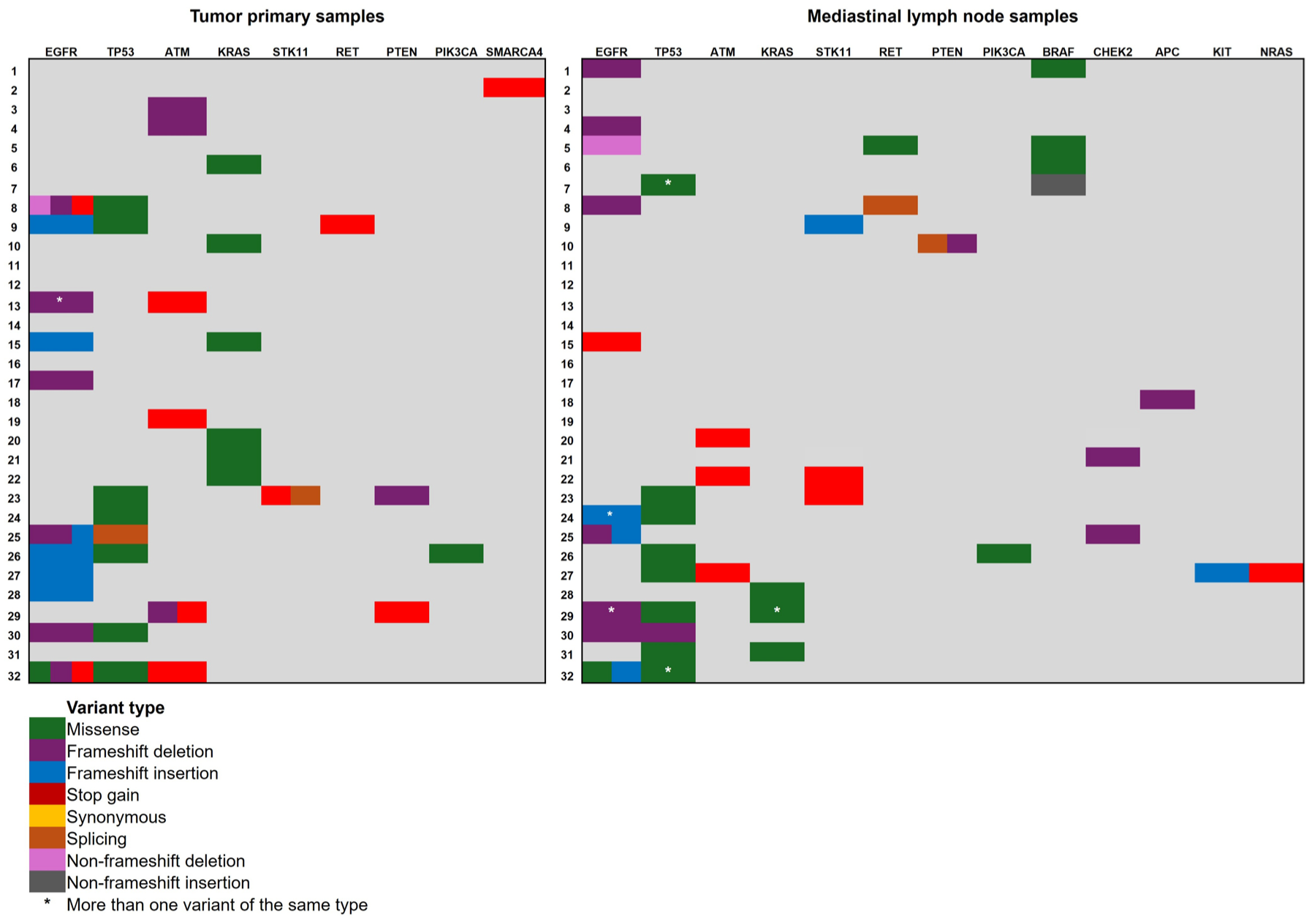
Distribution of genomic variants with strong and potential clinical significance (Tiers I and II) 28 is presented by gene and variant type, categorized by case (patient) and type of analyzed sample (PT and MLN). This figure demonstrates the discordance/concordance between PT and MLN genomic profiles and the presence of co-mutations.
Figure 1 illustrates the somatic variants of strong clinical significance found in the genes of the PT samples and corresponding MLN. The different types of variants are represented by different colors to obtain an overview of all variants observed in the study.
In PT samples, the EGFR gene harbored the highest proportion of clinically significant variants per patient, which were identified in 39.3% of cases (n = 11), followed by TP53 (28.6%, n = 8) and KRAS and ATM, each with 21.4% (n = 6). Variants were also observed at lower frequencies in PTEN (7.1%, n = 2) and in RET, PIK3CA, STK11, and SMARCA4, each accounting for 3.6% (n = 1) of the detected variants. For these calculations, we considered 28 patients, since 4 patients did not exhibit clinically significant variants.
Table 2 provides a detailed distribution of the total number of variants detected across the study, which are stratified by (a) histological group: Group A (MLN−) versus Group B (MLN+); (b) sample type: PT versus MLN.
Number of variants of strong and/or potential clinical significance found in groups A and B, by sample gene, and sample type.

Gene that presented the same variant in more than one patient.
Genes that presented the same variant in PT and MLN samples.
−, negative; +, positive; MLN, mediastinal lymph node; PT, primary tumor.
In the MLN samples in groups A and B, the EGFR gene remained the most frequently mutated gene, with clinically significant variants detected in 35.7% of the patients (n = 10); this was closely followed by TP53, whose variants were observed in 32.1% of patients (n = 9). Variants in KRAS and BRAF were observed in 14.3% (n = 4) of these genes, and ATM in 10.7% (n = 3) of patients. Additional genes with lower-frequency alterations included CHEK2 and RET, whose variants were each observed in 7.1% of the samples (n = 2), and STK11, whose variants appeared in 10.7% (n = 3). Single occurrences (3.6%, n = 1) of variants were also detected in PIK3CA, PTEN, NRAS, APC, and KIT.
KRAS mutations were identified in eight patients, and the hotspot variant c.34G>T (rs121913530) was present in six patients, namely, four in PT samples and two in MLN samples. Additionally, the variants c.35G>T and c.35G>A (rs121913529) were detected in three patients: two in PT samples and one in an MLN sample, where the latter was found along with the c.34G>T mutation. These findings highlight the recurrence of KRAS codon 12 alterations in PT and MLN tissues (see Supplemental Tables 1 and 2 for the full variant distributions).
Concomitant variant analysis (co-mutations)
An analysis of co-occurring genomic alterations revealed that 31.3% of patients (n = 10) had more than one variant in the same gene or across different genes within PT samples. Among these patients, 80.0% (n = 8) involved EGFR, with the most frequent combination being the EGFR and TP53 co-mutations, which were observed in 60.0% (n = 6) of the cases. Only one patient, who presented combined ATM/PTEN variants, exhibited co-mutations that did not involve EGFR or TP53.
Among the MLN samples, 43.8% of the patients (n = 14) harbored concomitant variants, and of these, TP53 was implicated in 64.3% (n = 9) of these cases, followed by EGFR in 57.2% (n = 8).
Covariation between EGFR and TP53 was present in 28.6% (n = 4) of the patients. Only one MLN sample and one PT sample showed covariation outside the EGFR/TP53 axis, specifically in ATM/STK11.
According to pairwise comparisons between the PT and MLN samples, 53.1% of patients (n = 17) had co-mutations in either or both tissues. Among these patients, 18.6% (n = 6) exhibited shared variants in both PT and MLN samples. Notably, this concordance was observed exclusively in MLN+ patients. No overlapping variants were identified between PT and MLN in the MLN– group, thus suggesting that variant burden and genomic complexity increase in lymph nodes once metastasis is established.
Of the PT samples, 15.7% (n = 5) presented no variants of interest; however, their corresponding MLN samples harbored clinically significant mutations, which suggests possible tumor heterogeneity or clonal evolution in lymphatic dissemination. Conversely, 15.7% of MLN samples (n = 5) contained no detectable variants; all of these cases belonged to the MLN– group. In contrast, every MLN+ sample harbored at least one variant, which supports the hypothesis that tumor cell clones carrying specific mutations are transferred to lymph nodes during metastasis.
Notably, 45.0% (n = 12) of MLN– samples nonetheless carried Tier I or Tier II variants, as classified by AMP/ASCO/CAP guidelines, 28 including documented clinical evidence and potential therapeutic implications. These findings reinforce the clinical utility of genomic profile even in histologically negative MLN, as they may inform treatment decisions and predict disease progression.
To assess the impact of genomic alterations on patient outcomes, OS was first evaluated via the Kaplan–Meier method. Analyses were performed separately for the PT and MLN samples, whereas patients were grouped collectively for OS analysis, regardless of their MLN histology status.
No statistically significant association was observed between the presence of clinically significant EGFR variants and OS. Among those with variants in PT samples, patients with wild-type EGFR had a median OS of 54 months, whereas those harboring EGFR mutations had a median OS of 52 months (p = 0.949). Similarly, among those with variants in MLN samples, patients with wild-type EGFR exhibited a median OS of 56 months, whereas those with EGFR mutations exhibited a median OS of 46 months (p = 0.342; Supplemental Figure 2(A) and (B)).
For the TP53 gene, although patients whose PT samples contained TP53 variants tended to be characterized by shorter survival times (median OS: 38 vs 57 months for wild-type samples), this difference did not reach the level of statistical significance (p = 0.247; Supplemental Figure 2(A)). However, the presence of TP53 variants in MLN samples was significantly associated with reduced OS (p = 0.001). Patients who harbored TP53 mutations in MLN exhibited a median OS of 26 months, whereas those with wild-type TP53 in MLN exhibited a median OS of 62 months (Figure 2(A)).

Survival curves for patients with variants in MLN samples. (a) TP53 variants in both study groups. (b) ATM gene variants in both study groups. These curves demonstrate that the presence of the variant negatively impacts the OS of patients with LUAD.
No significant differences in OS were detected among patients whose PT samples contained variants in the ATM gene (p = 0.260), with median OS values of 66 months for patients with ATM mutations and 50 months for patients with wild-type ATM (Supplemental Figure 2(B)). In contrast, patients whose MLN samples contained ATM variants were significantly more likely to receive a poorer prognosis (p = 0.036). Patients with wild-type ATM exhibited a median OS of 57 months, whereas those with mutations in ATM exhibited a significantly shorter OS of 30 months (Figure 2(b)).
Considering the co-mutation analysis, no significant associations were detected between OS and the presence of co-mutations in paired PT samples (PT/PT; p = 0.880) or between paired PT samples and MLN samples (PT/MLN; p = 0.399), as shown in Supplemental Figure 2(A) and (B). However, when MLN samples (MLN/MLN) were analyzed exclusively, a significant reduction in OS was observed in patients who harbored co-mutations. Patients with concomitant variants in MLN exhibited a median OS of 40 months, whereas patients without co-mutations exhibited a median OS of 67 months (p = 0.005; Figure 2). These findings suggest that the presence of multiple concurrent mutations in MLN tissue is associated with a worse prognosis. Figure 3 also shows the occurrence of co-mutations in the cohort analyzed in the study.

Survival curves for patients with covariants present in MLN samples (MLN/MLN). Notably, TP53 was the most frequently involved gene in these cases, as variants in TP53 were observed in 64.3% of MLN samples with concomitant alterations. This finding supports the hypothesis that co-mutations, particularly those that involve tumor suppressor pathways, may reflect more aggressive tumor behavior and an increased likelihood of systemic dissemination.
In a next step, we performed a COX univariate analysis and observed worse OS for patients with MLN histologically negative for tumor cells (p = 0.001) and harboring MLN co-mutation (p = 0.007). In our multivariate analysis, the best mathematical model (χ2 = 22.19; p < 0.001) demonstrated that OS was independently influenced by the age (p = 0.04), sex (p = 0.04), smoking status (p = 0.04), and lymph node metastasis (p = 0.05). In this multivariate model, co-mutation in MLN was included as a covariate to improve the model’s accuracy and its effects on OS (Table 3).
Factors associated with mortality risk in non-small cell carcinoma patients were analyzed using univariate and multivariate Cox regression analysis (n = 32).

Univariate analysis was performed without any adjustment to generate HRs with CIs for the individual risk of each survival parameter. Multivariate analysis was performed to analyze the effects of various risk parameters on survival. Bold values represent statistical significance. χ2 = 22.19; p < 0.001.
In accordance with the 7th Edition of the International Association for the Study of Lung Cancer (Ref. 20).
CI, confidence interval; HR, hazard ratio (β coefficient); MLN, mediastinal lymph nodes; PT, primary tumor.
The concordance and discordance rates between the genomic profiles of PT and MLN were calculated based on 28 patients, as 4 patients in the study were not exhibit clinically significant variants. Patients whose PT samples presented the same variant as MLN matched sample were considered concordant. Those patients with distinct variants in the PT and MLN samples were considered discordant. We observed that 17.8% (n = 5) of patients presented concordance between their PT and MLN profiles, a percentage that corresponded to patients with MLN+. Discordance between genomic profiles occurred in 82.2% (n = 23) of patients. Figure 1 shows the discordance between the detailed profiles.
Discussion
In this genomic study, we analyzed paired samples of LUAD and mediastinal MLN obtained via EBUS-TBNA from Brazilian patients. We aimed to identify clinically relevant variants and evaluate their associations with OS. NGS revealed a diverse profile of PT and MLN, with differences in variant distribution and prognostic implications.
In light of the paired profile, a relevant observation was the low concordance of somatic variants between PT and MLN in Group A (MLN−). Group B (MLN+) showed 54.5% concordance, suggesting that clonal dissemination from the PT to MLN becomes more evident as histologically detectable metastasis develops. These findings support the notion that genomic similarity increases once morphological metastasis is present, reflecting advanced tumor evolution.
Our results partially align with a recently reported concordance rate of 76.4% among a Chinese NSCLC cohort, 33 which is higher than that observed in our Brazilian patients. Differences in sequencing strategies, ethnic and environmental backgrounds, and clinical staging may explain this discrepancy. Other authors reported complete PT/MLN concordance by multiregion sequencing 34 ; however, they used a more comprehensive approach than the single-region EBUS-TBNA biopsies in our study. Our lower concordance likely reflects technical limitations and biological heterogeneity, including early subclonal MLN dissemination and limited tumor cellularity in histologically negative samples.
Importantly, we identified clinically significant variants even in MLN−, as 70.6% harbored Tier I or II variants in this group A, supporting the hypothesis that molecular changes may precede detectable morphological alterations. This highlights the potential clinical value of molecular profile in histologically negative nodes, which could serve as early markers of occult metastasis and inform risk stratification and treatment selection, especially when PT biopsies are unavailable or insufficient.
The most frequently altered genes across both sample types in our study included EGFR, TP53, KRAS, and ATM. These findings align with global LUAD data and highlight regional patterns. 35 Recent studies in China, 36 Romania, 37 and Korea 38 illustrate genomic diversity and support the limited concordance between liquid and tissue biopsies in early disease. Despite these variations, TP53 and EGFR remain central drivers in LUAD pathogenesis.
To our knowledge, this study is one of the first to characterize genomic alterations in EBUS-TBNA-derived MLN samples from Brazilian patients, highlighting the feasibility and clinical utility of minimally invasive techniques for genomic profile. Previous studies have demonstrated discordances between PT and MLN genotypes, particularly in EGFR, with therapeutic implications.39,40 For instance, patients with EGFR variants in MLN but not in the PT respond favorably to EGFR-TKIs, demonstrating that MLN metastatic sites may harbor actionable variants that influence treatment selection and would be missed if only the PT is analyzed.
ATM variants, which can affect prognosis and treatment response in NSCLC,17,41,42 were observed in our cohort, reinforcing their potential as prognostic biomarker. Deficiency of the ATM protein has been observed in up to 40% of LUADs, making it a potential biomarker and therapeutic target for NSCLC. 43 Co-occurring mutations, such as TP53–EGFR co-mutations, were associated with worse OS, highlighting the importance of comprehensive molecular profile beyond single-gene evaluation.44,45
Our findings also suggest the clinical sampling strategy. When both PT and MLN are accessible, MLN should be prioritized in patients with nodal involvement, as they may harbor clinically significant variants that are not detected in PT. However, PT sampling remains essential, particularly when MLN are inaccessible, or there is no nodal disease. When feasible, a complementary approach integrating the molecular profile of both sites provides a comprehensive genomic landscape, supporting personalized therapy and accurate prognostic assessment. Procedure risks, sample quality, and tumor cellularity should guide site selection.
Interestingly, TP53 variants in PT were not associated with worse survival, contrasting with other reports.40,46 However, TP53 variants in MLN, regardless of histological classification, were significantly associated with poorer OS, emphasizing the prognostic value of MLN profile. This finding may reflect biological differences between PTs and metastatic nodes. MLN metastases could represent more aggressive subclones of the PT, enriched for TP53 alterations that drive adverse outcomes. 47 In addition, intratumoral heterogeneity may result in a differential distribution of key variants between PT and MLN, such that the prognostic impact of TP53 variants was more evident in metastatic nodes. Methodological factors may also contribute to these findings; the small number of MLN-positive cases may have influenced the results, compared with larger cohorts in prior studies. Differences in variant detection platforms, criteria for defining clinically significant variants, and follow-up duration may also account for divergent findings. Furthermore, single-region of PT biopsies may miss subclonal TP53 variants that were captured in MLN, highlighting the importance of sampling both sites for comprehensive molecular assessment. Taken together, these biological and technical considerations support the need to extend molecular profile to MLN to improve prognostic accuracy and guide personalized therapeutic strategies. Although our results suggest a potential prognostic role for TP53, ATM, and co-mutations in MLN, they should be interpreted cautiously, since larger prospective studies are needed to validate these associations.
Nevertheless, regarding the OS analysis by a multivariate Cox regression model, we demonstrated that the worst prognosis was associated with patients who presented the following profile: young women, nonsmokers, with MLN histologically negative for metastasis, and harboring the MLN co-mutation.
We recognize that our study has limitations, such as the small sample size and the fact that it is a single-center design; however, it is the first Brazilian study to analyze paired PT and MLN samples using NGS. The absence of paired normal tissue or blood samples to exclude germline variants also constituted a significant limitation. Since this was a retrospective study, the number of patients included in the sample was limited by technical challenges encountered when working with long-term stored FFPE samples. Furthermore, 50% of the patients had already died by the time the study began. To overcome these issues, variants were manually confirmed in public databases (OncoKB, COSMIC, Varsome, and Franklin), in accordance with the 2017 AMP/ASCO/CAP guidelines for the classification and interpretation of somatic variants in cancer.
Our findings demonstrate that histologically negative MLN can anchorage clinically significant somatic variants, particularly in TP53 and ATM, which are associated with lower OS. Partial genomic concordance between PT and MLN, especially in histologically positive nodes, reflects tumor evolution and metastasis dynamics. These results support the proposal to integrate MLN genomic profile into routine diagnostic workflows, enhancing the identification of occult metastases, and complementing PT analysis for a more comprehensive genomic assessment.
Concerning KRAS, we observed the G12C variant in both PT and MLN, which is consistent with the association of this variant with smoking and aggressive tumor biology. However, this mutation did not significantly affect survival, echoing the results of previous studies that have also reported no correlation between KRAS status and metastasis or prognosis. 48 These findings suggest that while KRAS is biologically relevant, other genes, such as TP53 and ATM, may have greater prognostic and therapeutic significance in the context of NSCLC.
In the current study, we observed discrepancies in EGFR variant status between PT and MLN. These findings highlight the potential clinical and therapeutic implications of genotyping MLN and metastatic sites. Patients with EGFR variants detected in MLN but not in PT may benefit from EGFR-targeted TKIs, emphasizing the importance of including metastatic sites in the molecular analysis. Conversely, to evaluate the genomic profile only from the PT may result in under-treatment or in conventional therapy for patients who presented EGFR variants in MLN. Previous studies have reported similar patterns of EGFR discordance between PT and metastatic sites. There is evidence that patients with EGFR variants in metastatic MLN respond favorably to EGFR-TKI therapy, even when the PT is of wild-type.1,2 These results corroborate the translational relevance of PT and MLN assessment in clinical decision-making. Prospective studies that integrate a matched genomic profile with treatment follow-up are required to assess the full impact of EGFR discordance on clinical outcomes.
Conclusion
This study represents the first comprehensive comparative analysis of clinically significant somatic variants in matched PT and MLN obtained via EBUS-TBNA from Brazilian patients with LUAD. These findings underscore the critical importance of conducting population-based genomic studies, which are essential for elucidating tumor heterogeneity, improving staging precision, identifying robust prognostic biomarkers specific to regional genetic backgrounds, and advancing the implementation of precision oncology in LC management.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251409007 – Supplemental material for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma
Supplemental material, sj-docx-1-tam-10.1177_17588359251409007 for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma by Caroline Silvério Faria, Camila Machado Baldavira, Tabatha Gutierrez Prieto, Ilka Lopes Santoro, Viviane Rossi Figueiredo, Ellen Caroline Toledo do Nascimento, Leslie Domenici Kulikowski, Teresa Yae Takagaki, Murilo Cervato, Ricardo Mingarini Terra, Vera Luiza Capelozzi and Leila Antonangelo in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251409007 – Supplemental material for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma
Supplemental material, sj-docx-2-tam-10.1177_17588359251409007 for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma by Caroline Silvério Faria, Camila Machado Baldavira, Tabatha Gutierrez Prieto, Ilka Lopes Santoro, Viviane Rossi Figueiredo, Ellen Caroline Toledo do Nascimento, Leslie Domenici Kulikowski, Teresa Yae Takagaki, Murilo Cervato, Ricardo Mingarini Terra, Vera Luiza Capelozzi and Leila Antonangelo in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-5-tam-10.1177_17588359251409007 – Supplemental material for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma
Supplemental material, sj-docx-5-tam-10.1177_17588359251409007 for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma by Caroline Silvério Faria, Camila Machado Baldavira, Tabatha Gutierrez Prieto, Ilka Lopes Santoro, Viviane Rossi Figueiredo, Ellen Caroline Toledo do Nascimento, Leslie Domenici Kulikowski, Teresa Yae Takagaki, Murilo Cervato, Ricardo Mingarini Terra, Vera Luiza Capelozzi and Leila Antonangelo in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-jpg-3-tam-10.1177_17588359251409007 – Supplemental material for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma
Supplemental material, sj-jpg-3-tam-10.1177_17588359251409007 for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma by Caroline Silvério Faria, Camila Machado Baldavira, Tabatha Gutierrez Prieto, Ilka Lopes Santoro, Viviane Rossi Figueiredo, Ellen Caroline Toledo do Nascimento, Leslie Domenici Kulikowski, Teresa Yae Takagaki, Murilo Cervato, Ricardo Mingarini Terra, Vera Luiza Capelozzi and Leila Antonangelo in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-jpg-4-tam-10.1177_17588359251409007 – Supplemental material for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma
Supplemental material, sj-jpg-4-tam-10.1177_17588359251409007 for Clinically relevant somatic variants and genomic discordance between primary tumors and mediastinal lymph nodes in lung adenocarcinoma by Caroline Silvério Faria, Camila Machado Baldavira, Tabatha Gutierrez Prieto, Ilka Lopes Santoro, Viviane Rossi Figueiredo, Ellen Caroline Toledo do Nascimento, Leslie Domenici Kulikowski, Teresa Yae Takagaki, Murilo Cervato, Ricardo Mingarini Terra, Vera Luiza Capelozzi and Leila Antonangelo in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We are grateful to the staff of the Histopathology Laboratory of FMUSP, the Pathological Anatomy Laboratory of Hospital Sírio Libanês, the Pathology Laboratory of ICESP, Amom Mendes Nascimento from the Cytogenomics Laboratory of LIM 03, the Large Scale Sequencing Laboratory of FMUSP (SELA-FMUSP), Varsomics, and AJE for the idiomatic review.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.