
Abstract
Background:
Next-generation sequencing (NGS) of plasma circulating tumor DNA (ctDNA) shows promise as a minimally invasive alternative to tissue sequencing. However, concordance between genomic alterations in tumor tissue and plasma ctDNA remains incompletely characterized in ovarian cancer, particularly in the pretreatment setting.
Objectives:
To identify factors influencing concordance between tissue and ctDNA genomic profiling and explore the potential clinical implications (including treatment and survival outcomes) of this concordance.
Design:
A prospective, single-center, observational study.
Methods:
A total of 40 matched pretreatment tumor specimens and blood samples were prospectively collected from patients with ovarian cancer and subsequently sequenced using a customized gene panel. Overall and individual concordance rates were calculated as the ratio of total concordant mutations to total tissue mutations, with patients stratified into highly concordant (⩾50%) and poorly concordant (<50%) groups.
Results:
The overall tissue–plasma concordance rate was 40.8%, with shared variants exhibiting identical abundance patterns across sample types and capturing the majority of functionally relevant mutations. Single-nucleotide variants in tissue showed a higher detection rate in plasma than structural variants. Individual concordance rates displayed significant inter-patient variability. Higher tissue tumor mutation burden (odds ratio (OR) 2.165, 95% confidence interval (CI) 1.183–3.965) and plasma ctDNA fraction (OR 1.433, 95% CI 1.063–1.933) were independently associated with high concordance rates. In advanced-stage patients, the poorly concordant group showed lower CA125 elimination rate constant K (KELIM) scores (median 0.7 vs 1.3; 15.0% vs 68.8% patients with score ⩾1), indicating reduced chemosensitivity. The poorly concordant group demonstrated a higher disease recurrence rate (40.0% vs 6.2%) and elevated early recurrence risk (12-month progression-free survival rate 82.0% vs 100.0%) compared with the highly concordant group.
Conclusion:
In the field of ovarian cancer, NGS of ctDNA showed moderate concordance with tissue-based sequencing in the pretreatment setting, influenced by both technical and biological factors. The tissue–plasma concordance may serve as a chemosensitivity and prognostic indicator.
Keywords
Introduction
Ovarian cancer is the leading cause of death among gynecological malignancies, often presenting in advanced stages at the time of diagnosis. 1 Cytoreductive surgery followed by platinum-based chemotherapy is the standard of care. Despite the implementation of anti-angiogenic treatment with bevacizumab and poly ADP-ribose polymerase (PARP) inhibitors, the overall prognosis of advanced ovarian cancer remains poor with the majority of patients experiencing recurrence. 2 Along with the progress of drug development, novel targeted agents have been proved to have promising clinical activity, which requires the precise detection of tumor molecular features to guide treatment decision-making. 3
In recent years, the rapid development of next-generation sequencing (NGS) technology has made it feasible for clinical application. Traditionally, NGS is performed on tissue samples for molecular genetic profiling to guide targeted therapy in clinical practice. However, tissue biopsies are not only invasive but often challenging to obtain, particularly given the characteristic peritoneal dissemination pattern of advanced ovarian cancer, which introduces significant spatial heterogeneity across different metastatic sites. 4 To overcome these limitations, minimally invasive liquid biopsy through plasma sampling has emerged as a promising alternative, which allows a more holistic view on tumor molecular genetic landscape.5,6 This approach, primarily based on circulating tumor DNA (ctDNA) detection, has demonstrated potential for the whole-course disease management in patients with ovarian cancer, including early diagnosis, minimal residual disease assessment, actionable target detection, and real-time treatment response monitoring. However, several inherent limitations of blood-based liquid biopsy in ovarian cancer should also be acknowledged. 7 First, sensitivity is compromised by low ctDNA concentrations in blood, particularly in early-stage disease, creating substantial technical challenges for reliable detection. Second, specificity concerns arise from the difficulty in distinguishing ovarian cancer-derived ctDNA from cell-free DNA (cfDNA) released by benign gynecologic conditions, inflammatory processes, or concurrent malignancies. Finally, the absence of standardized detection and quality control methods impedes meaningful comparison between studies and limits practical clinical implementation.
Biological limitations specific to ovarian cancer may also impact ctDNA detection and quantification. First, the genomic instability characteristic of ovarian cancer, driven by TP53 loss-of-function and homologous recombination deficiency (HRD), poses both technical and biological challenges for ctDNA-based genomic profiling. The extensive chromosomal alterations and complex structural variants (SVs) inherent to this genomic instability can be difficult to accurately detect and quantify in the low-concentration ctDNA found in plasma samples.8,9 Moreover, ovarian cancer, particularly the high-grade serous subtype, primarily spreads via transcoelomic seeding within the peritoneal cavity rather than through hematogenous dissemination. 10 This metastatic pattern significantly limits ctDNA shedding into systemic circulation, potentially resulting in lower plasma ctDNA concentrations compared to tumors that predominantly metastasize hematogenously.
Therefore, a critical concern in clinical practice is the concordance between genomic profiles derived from tumor tissue DNA and plasma ctDNA. 11 Previous systematic review has revealed highly variable tissue–plasma concordance rates in ovarian cancer, ranging from 25% to 100% across distinct sampling timepoints, detection methods, and gene panels, leading to ongoing debate in the field. 12 Despite the clinical importance of this issue, there remains a paucity of research examining the factors that influence tissue–plasma concordance, and the clinical implications of concordance levels are not well understood.
In this study, we systematically evaluated the concordance between genomic alterations detected in paired pretreatment tumor tissue and plasma samples from a prospectively enrolled cohort, aiming to identify the clinicopathological and tumor molecular factors influencing tissue–plasma concordance and investigate its potential clinical implications.
Methods
Patient enrollment and sample acquisition
A prospective collection of fresh tumor specimens and peripheral blood samples from patients with ovarian cancer was conducted at Peking Union Medical College Hospital between April 2023 and February 2025. All surgical procedures, systemic treatments, and follow-up care were performed by a single experienced gynecologic oncology team to ensure consistency in clinical management. Standard carboplatin (area under the curve (AUC) = 5)/paclitaxel (175 mg/m2) was administered every 3 weeks for neoadjuvant and adjuvant chemotherapy. During adjuvant chemotherapy, the regimens were given with or without bevacizumab, and upon completion, patients either received PARP inhibitors for maintenance therapy or not. Women were followed up and monitored by clinical examination, CA125 measurement, and imaging test (computed tomography scan or positron emission tomography-computed scan every 3 months). The data cutoff date ended in May 2025. It was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines, and approved by the Ethics Committee of Peking Union Medical College Hospital (approval number: I-24PJ1296). All participants provided written informed consent before enrollment. The reporting of this study conforms to the STROBE statement 13 (checklist in Supplemental File 1).
The inclusion criteria for the current study were as follows: (1) histologically confirmed non-mucinous epithelial ovarian cancer; (2) primary disease (without prior recurrence); and (3) availability of paired pretreatment tumor tissue and plasma samples. Clinicopathologic variables were recorded, including age, treatment strategy, outcome of cytoreduction, International Federation of Gynecology and Obstetrics (FIGO) stage, histologic subtype, largest tumor size, tumor Ki-67 index, and serial CA125 measurements. The CA125 elimination rate constant K (KELIM) score was calculated using the online tool (https://www.biomarker-kinetics.org/CA-125) developed by You et al. 14 to predict response to chemotherapy during postoperative adjuvant chemotherapy after primary debulking surgery (PDS) or neoadjuvant chemotherapy (NACT). A higher KELIM score indicates faster CA125 elimination and greater chemosensitivity, with values ⩾1 considered as favorable scores. Progression-free survival (PFS) was defined as the time from stopping the last chemotherapy until the date of first disease recurrence. Assessment of disease recurrence was based on RECIST criteria or CA125 progression criteria as defined by the Gynecological Cancer InterGroup.
The pretreatment tumor specimens were obtained from PDS or diagnostic laparoscopy for patients designated to NACT. Fresh tissue samples were collected either from the primary sites (ovaries and fallopian tubes) or metastatic sites within the abdominal cavity, including the omentum and peritoneum. Matched pretreatment plasma samples were collected on the day of surgical procedures.
DNA extraction and targeted sequencing
Approximately 10 ml of venous blood was collected in a cfDNA storage tube, which was gently inverted and mixed. The subsequent process was conducted within 4 h after collection. Blood samples were centrifugation first at 800g for 15 min, followed by a second centrifugation at 1600g for 10 min. cfDNA in the double-spun plasma was extracted using QIAmp Circulating Nucleic Acid Kit (Qiagen, Duesseldorf, Germany). Genomic DNA of the white blood cells was extracted using DNeasy Blood & Tissue Kit (Qiagen, Duesseldorf, Germary) and used as germline controls. Fresh tissues were promptly placed in tissue preservation solution, and genomic DNA was extracted using the DNA FFPE Tissue Kit (Qiagen, Duesseldorf, Germany). All DNA samples were quantified by Qubit 3.0 using the dsDNA HS Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s recommendations.
Library preparations were performed with the KAPA Hyper Prep Kit (KAPA Biosystems, Wilmington, MA, USA) according to the manufacturer’s suggestions for different sample types. In brief, sequential operations of end-repairing, A-tailing, and indexed adapter ligation were performed to 11.2–164.8 ng (median: 42.9 ng) of cfDNA or 1–2 μg of fragmented genomic DNA, followed by size selection using Agencourt AMPure XP beads (Beckman Coulter, Miami, FL, USA). Hybridization-based target enrichment was carried out with a customized NGS panel of 2365 cancer-relevant genes (Shielding™ Ultra panel; Nanjing Geneseeq Technology, Nanjing, Jiangsu, China), and xGen Lockdown Hybridization and Wash Reagents Kit (Integrated DNA Technologies, Coralville, IA, USA). Captured libraries were amplified by PCR with Illumina p5 and p7 primers in KAPA HiFi HotStart ReadyMix (KAPA Biosystems, Wilmington, MA, USA), followed by quantification by qPCR using the KAPA Library Quantification Kit (KAPA Biosystems, Wilmington, MA, USA) and purification using Agencourt AMPure XP beads. Library fragment size was assessed by Bioanalyzer 2100 (Agilent Technologies, Santa Clara, CA, USA). Sequencing of the target-enriched library was then performed on the Illumina HiSeq4000 NGS platform. Targeted sequencing of the BRCA1 and BRCA2 genes was performed simultaneously on the tissue samples. The average coverage sequencing depth of the whole blood control was 9000×. The average sequencing depth was approximately 1000× and 30,000× for tissue and plasma, respectively.
DNA samples were analyzed through bioinformatic processing, including quality control, alignment to the reference human genome, deduplication, cross-sample contamination estimation, and variant discovery. A comprehensive description of this entire pipeline is provided in the supporting methods in Supplemental File 2. Briefly, somatic mutations (SMs) were called using VarScan2 15 and CNVkit (https://cnvkit.readthedocs.io) with a paired workflow. These mutations included both single-nucleotide variants (SNVs) and SVs. SVs included insertions, deletions, duplications, inversions, copy number variants (CNVs), and so on. 16 The phenotypic impact of variants was assessed using CADD 17 (score >10 indicating deleterious effects), SIFT 18 (classifying variants as tolerated or deleterious), and PolyPhen 19 (categorizing variants as benign, possibly damaging, or probably damaging). A series of custom filters was applied to the called mutations to remove false positives and increase specificity.
Plasma ctDNA fraction was estimated using the maximum variant allele frequency (VAF) among detected plasma ctDNA mutations. 20 Tissue tumor mutation burden (TMB) was counted by summing all base substitutions and indels in the coding region of targeted genes, including synonymous alterations to reduce sampling noise and excluding known driver mutations as they are over-represented in the panel, as previously described. 21 TMB-high cutoff was set as ⩾10 mutations per mega-base (Mb) according to the widely accepted cutoff measured by FoundationOne panel and validated in a few studies. 22 The HRD score was calculated based on the genome-wide allele-specific copy number result and composed of three parts: (1) loss of heterozygosity: the number of segments with ⩾15 Mb length (but not cover the whole chromosome), per segment minor copy number (mCN) = 0, and per segment total copy number (tCN) > 0; (2) telomeric allelic imbalance (TAI): the number of segments with allelic imbalance (mCN! = tCN − mCN) extend to the telomeric end of a chromosome; (3) large-scale state transitions, number of chromosomal breaks between adjacent segments of at least 10 Mb, with a distance between them not larger than 3 Mb. An HRD-positive threshold of 38 was applied, which had been previously validated in Chinese patients with HRD-associated cancers, including ovarian cancer. 23 The laboratory (Nanjing Geneseeq Technology, Nanjing, Jiangsu, China) is certified by the Clinical Laboratory Improvement Amendments, College of American Pathologists, and International Organization for Standardization (ISO15189).
Concordance in genomic alterations between tumor tissue and plasma ctDNA
Using tumor tissue DNA as the gold standard, concordance analysis was conducted to evaluate the mutation detection consistency between tumor tissue and plasma samples. The overall concordance rate was calculated as the ratio of total concordant mutations (detected in both tumor tissue and paired plasma ctDNA) to the total mutations identified in tumor tissue. For individual patients, the concordance rate was determined by dividing the number of shared mutations between paired tumor tissue and plasma samples by the total number of mutations found in the tumor tissue.
Identification of factors influencing tissue–plasma genomic alterations concordance
Previous studies in other solid tumors (gastric, lung, and prostate cancer) have demonstrated tissue–plasma concordance rates ranging from 40% to 50%, with rates exceeding 50% considered high concordance.24 –26 Therefore, patients in our study were classified into highly concordant (⩾50%) or poorly concordant (<50%) groups based on their individual concordance rates. Univariate logistic regression was initially performed on clinicopathological variables and tumor molecular characteristics to identify potential predictors of high tissue–plasma concordance rates (⩾50%). Variables with p ⩽ 0.1 in univariate analysis were subsequently entered into a multivariate logistic regression model. The selection of p ⩽ 0.1 as the inclusion criterion follows established conventions to avoid excluding potentially relevant variables. 27 Model calibration was evaluated using the Hosmer–Lemeshow goodness-of-fit test.
Receiver operating characteristic curves were constructed to evaluate the discriminatory performance of significant variables from the multivariate logistic regression model for predicting high concordance rates. AUC with 95% confidence intervals (CIs) was calculated using the trapezoidal rule. Optimal cutoff values were determined using the Youden index, which maximizes the sum of sensitivity and specificity. These cutoff values were used to convert continuous variables into binary predictors, and their performance was re-evaluated to assess threshold robustness.
Statistical analysis
Quantitative variables were presented as medians with interquartile ranges, while categorical variables were expressed as numbers and percentages. Between-group comparisons were performed using Student’s t test, Fisher’s exact test, or the nonparametric Mann–Whitney U test as appropriate. Correlations were assessed using Spearman’s rank correlation analysis. PFS was analyzed using the Kaplan–Meier method, with differences between curves evaluated by the log-rank test. Statistical analyses and visualization were performed using the R system (version 4.0.0; Posit Software, Boston, MA, USA), SPSS software (version 29.0; IBM, Armonk, NY, USA), and GraphPad Prism (version 10.0; Graphpad Software, San Diego, CA, USA). All statistical tests were two-sided, with p < 0.05 considered statistically significant.
Results
Patient characteristics
Between April 2023 and February 2025, we prospectively screened 113 patients with suspected ovarian malignancy. Following exclusion criteria, 40 patients with paired pretreatment tumor tissue and plasma samples were included in the final analysis. Patient selection and enrollment are detailed in Figure 1. The clinicopathologic characteristics of the study cohort are summarized in Table 1. All patients received standard-of-care treatment. The majority of patients presented with advanced-stage disease (90.0%, 36/40) and high-grade serous histology subtype (92.5%, 37/40). PDS was performed in 62.5% (25/40) of patients, while 37.5% (15/40) received NACT. Of the 15 patients receiving NACT, one patient did not proceed to interval debulking surgery because of poor performance status. The median follow-up time was 16.6 months (range: 3.8–26 months), with 70.0% (28/40) of patients followed for more than 12 months.
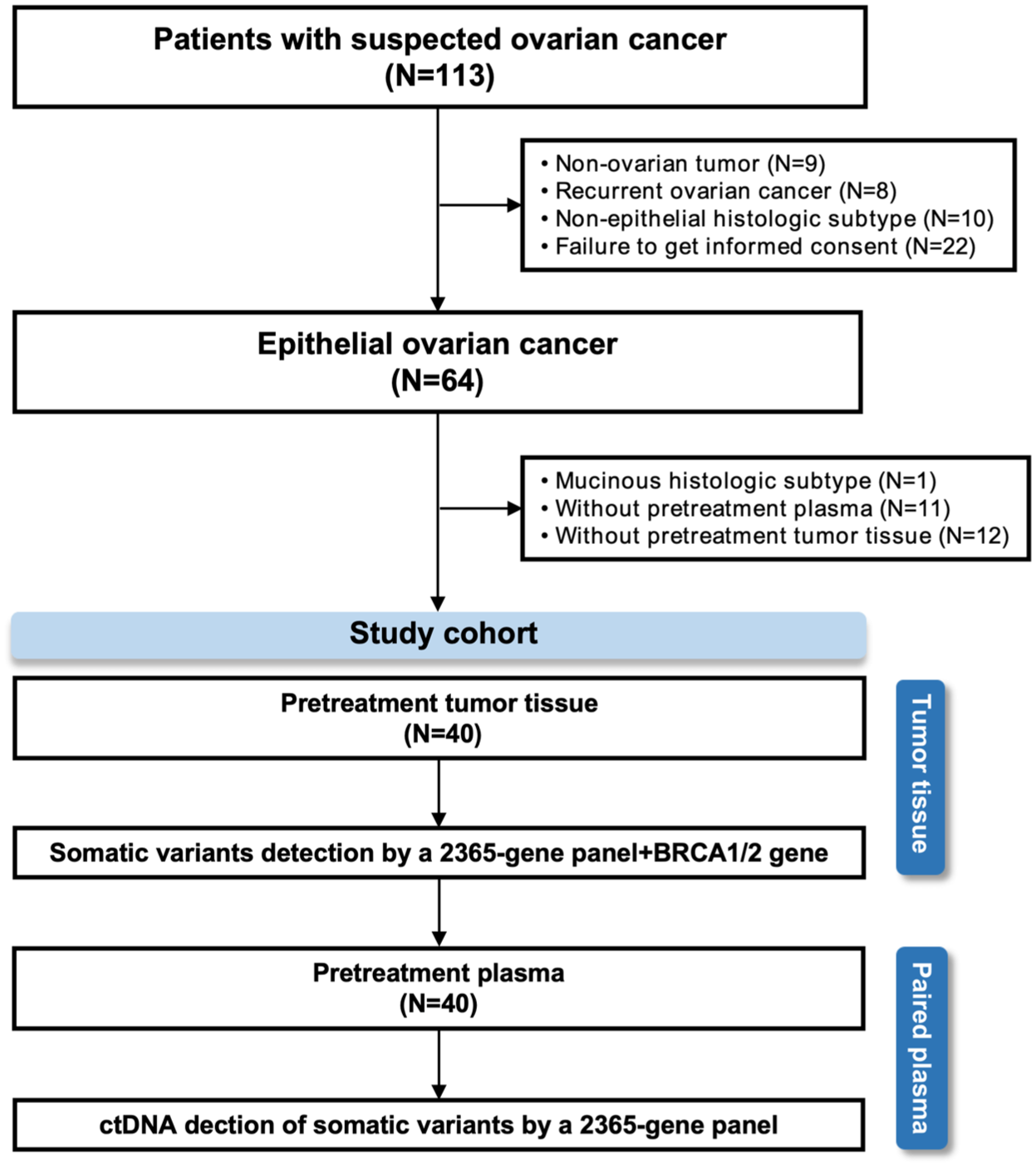
Patient enrollment and sample collection flowchart.
Baseline characteristics of patients in the poorly and highly concordant groups.
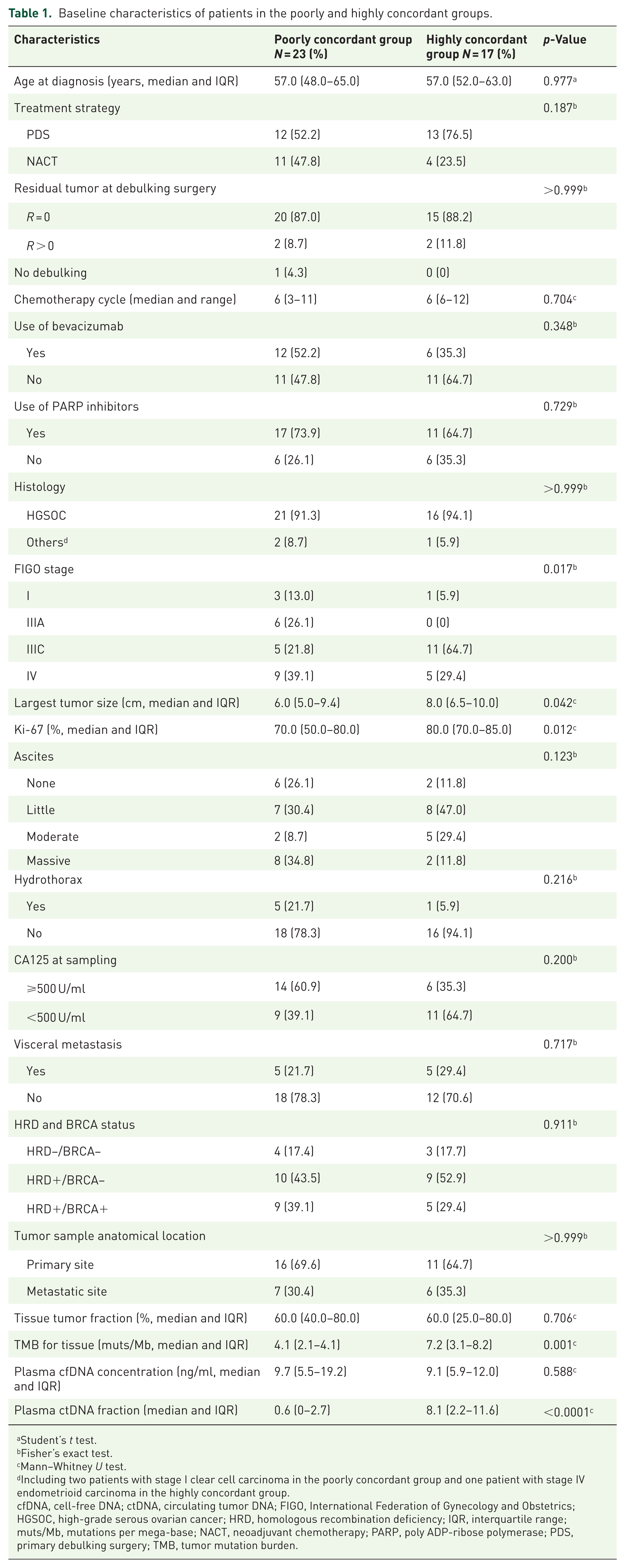
Student’s t test.
Fisher’s exact test.
Mann–Whitney U test.
Including two patients with stage I clear cell carcinoma in the poorly concordant group and one patient with stage IV endometrioid carcinoma in the highly concordant group.
cfDNA, cell-free DNA; ctDNA, circulating tumor DNA; FIGO, International Federation of Gynecology and Obstetrics; HGSOC, high-grade serous ovarian cancer; HRD, homologous recombination deficiency; IQR, interquartile range; muts/Mb, mutations per mega-base; NACT, neoadjuvant chemotherapy; PARP, poly ADP-ribose polymerase; PDS, primary debulking surgery; TMB, tumor mutation burden.
Overall, somatic alterations were detected in all tumor tissues and in 31 (77.5%) of the plasma samples. Among the nine cases where no mutations were detected in plasma, three patients had stage I disease, four had stage IIIA disease, and two had stage IV disease. Notably, they were also factored into subsequent concordance analyses.
Mutational concordance in paired pretreatment tissue and plasma and its associations with clinicopathologic factors
The landscape of somatic genomic alterations detected in tumor tissue and plasma ctDNA is illustrated in Figure 2. In total, 353 non-synonymous SMs were detected in pretreatment tumor tissues, with an average of 8.8 SMs per case (range: 2–21). SNVs constituted the majority of SMs (76.8%, N = 271), while SVs accounted for the remaining 23.2% (N = 82). The most frequently mutated genes were TP53 (92.5%), followed by KRAS (17.5%), SOX2 (15.0%), and MYC and PIK3CA (both 12.5%). Analysis of plasma ctDNA revealed 220 non-synonymous SMs, comprising 89.5% SNVs (N = 197) and 10.5% SVs (N = 23). TP53 was the most frequently mutated gene in plasma ctDNA, occurring in 87.1% of cases.

Genomic landscape with clinicopathological features in enrolled patients: tumor tissue (N = 40) and matched plasma ctDNA (N = 31, excluding nine plasma-negative cases).
Across all 40 patients, 144 SMs were concordant between tumor tissue and ctDNA genomic profiles, 209 SMs were exclusive to tumor tissue, and 76 SMs were detected solely in plasma ctDNA. The overall concordance rate was 40.8%. Notably, tissue-specific mutations undetected in plasma samples demonstrated lower VAF than shared mutations (median VAF 4.2% vs 28.7%, p < 0.0001, Mann–Whitney U test). A weak but statistically significant correlation was observed between the VAF of shared mutations in tumor tissue and plasma samples (r = 0.3099, p = 0.0002, Spearman’s rank correlation, Figure S1 in Supplemental File 3). The concordance rate for SNVs was 46.5% (126/271), higher than that for SVs at 22.0% (18/82; Figure 3(a), left and middle). Between two major SV subtypes, deletions showed a concordance rate of 30.6% (11/36), while CNVs had a lower rate of 11.4% (4/35; Figure 3(a), right). Furthermore, shared mutations showed significantly higher proportions of functionally relevant alterations compared to non-shared mutations (CADD score >10: 70.8% vs 55.1%, p = 0.002; SIFT deleterious: 46.5% vs 35.8%, p = 0.032; PolyPhen probably and possibly damaging: 45.8% vs 35.4%, p = 0.037; Chi-square test, Figure 3(b)), which demonstrated that plasma ctDNA successfully identified most driver mutations in tumor tissue.

Concordance analysis of tumor tissue and plasma ctDNA mutation profiles. (a) Overall mutation distribution showing tumor tissue-only, ctDNA-only, and concordant variants by mutation type. (b) Functionally relevant mutation distribution between shared and non-shared variants based on three computational prediction approaches.
Regarding the concordance at the individual patient level, tissue-based concordance rates ranged from 0 to 83.3% (median 40.0%), with 42.5% of patients (17/40) showing rates ⩾50% and 57.5% of patients (23/40) showing rates <50% (Figure 4(a)). Patients were stratified into a poorly concordant group (concordance rate <50%) and a highly concordant group (concordance rate ⩾50%). As demonstrated in Table 1, a statistically significant discrepancy was observed between the two groups regarding the baseline clinicopathological factors, including FIGO stage, largest tumor size, tumor Ki-67 index, and TMB for tissue and plasma ctDNA fraction. By contrast, no significant differences were observed between the poorly and highly concordant groups with respect to HRD and BRCA status (p = 0.911). The distribution of HRD−/BRCA−, HRD+/BRCA−, and HRD+/BRCA+ categories was similar between the two concordance groups. Subsequent univariate and multivariate logistic regression analyses linked having a high concordance rate with increased tissue TMB (odds ratio (OR) 2.165, 95% CI 1.183–3.965) and plasma ctDNA fraction (OR 1.433, 95% CI 1.063–1.933; Table 2). The multivariate model showed good calibration as assessed by the Hosmer–Lemeshow test (χ2 = 3.333, df = 8, p = 0.912). When analyzed as continuous variables, the combination of tissue TMB and plasma ctDNA fraction demonstrated superior predictive performance (AUC 0.913, 95% CI 0.823–1.000) compared with either plasma ctDNA fraction alone (AUC 0.855, 95% CI 0.742–0.969) or tissue TMB alone (AUC 0.786, 95% CI 0.629–0.944; Figure 4(b)). The optimal cutoffs for predicting concordance rate ⩾50% were established at 4.6 muts/Mb for tissue TMB and 0.7% for plasma ctDNA fraction. These metrics maintained comparable predictive value when analyzed as binary variables (Figure 4(c)).
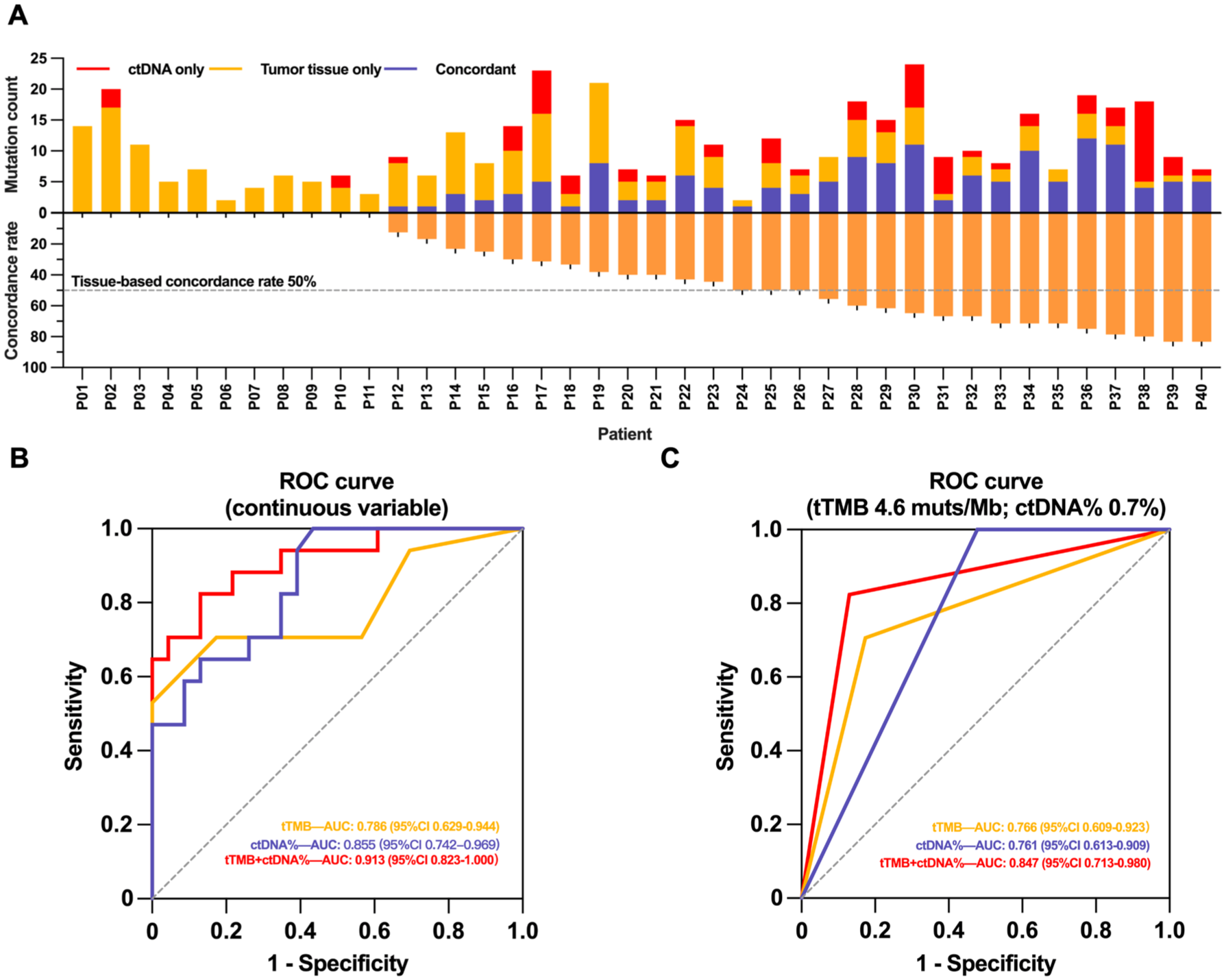
Individual concordance rates and predictive factors. (a) Patient-level mutation distribution and corresponding tissue–plasma concordance rates. (b, c) ROC curve analysis demonstrating predictive performance of tTMB and plasma ctDNA fraction for identifying high individual concordance rates (⩾50%).
Univariate and multivariate logistic analyses of factors predicting high tissue–plasma concordance rate (⩾50%).

cfDNA, cell-free DNA; CI, confidence interval; ctDNA, circulating tumor DNA; FIGO, International Federation of Gynecology and Obstetrics; HGSOC, high-grade serous ovarian cancer; HRD, homologous recombination deficiency; OR, odds ratio; TMB, tumor mutation burden.
Clinical implications of mutational concordance in paired pretreatment tissue and plasma
Given that patients with early-stage ovarian cancer exhibit fundamentally different clinical behavior and more favorable treatment and survival outcomes compared to those with advanced disease, and their limited ctDNA shedding is likely attributable to restricted lesion extent rather than tumor biological factors, subsequent analyses were restricted to advanced-stage (FIGO stage III–IV) patients (N = 36, including 35 patients with high-grade serous ovarian cancer and 1 patient with endometrioid carcinoma) to avoid potential confounding effects.
KELIM scores were calculated for all 36 advanced-stage patients. The advanced-stage patients in the poorly concordance group had a significantly lower KELIM score compared with the highly concordance group, whether analyzed as a continuous variable (median 0.7 vs 1.3, p < 0.0001, Student’s t test, Figure S2(A) in Supplemental File 3) or a binary variable (15.0% vs 68.8% of patients with KELIM score ⩾1, p = 0.002, Fisher’s exact test, Figure S2(B) in Supplemental File 3), suggesting reduced chemosensitivity as indicated by a slower CA125 elimination rate. A positive correlation was observed between individual concordance rates and KELIM scores (r = 0.7019, p < 0.0001, Spearman’s rank correlation, Figure 5(a)). At data cutoff, advanced-stage patients in the poorly concordant group demonstrated a higher proportion of disease recurrence compared with those in the highly concordant group (40.0% vs 6.2%, p = 0.026, Fisher’s exact test, Figure 5(b)). Further survival analysis of 12-month PFS rates between groups showed that patients in the poorly concordant group had higher early recurrence risk compared with those in the highly concordant group (12-month PFS rate 82.0% vs 100.0%). In addition, median PFS was shorter in the poorly concordant group (18.2 months vs unreached, p = 0.027; Figure 5(c)).

Clinical implications of tissue–plasma concordance in patients with advanced-stage ovarian cancer (N = 36). (a) Correlation between individual tissue–plasma concordance rates and chemosensitivity marker KELIM scores. (b) Disease recurrence rates between poorly and highly concordant groups. (c) PFS analysis comparing poorly versus highly concordant groups.
Discussion
In this prospective study, we systematically evaluated the concordance between pretreatment tumor tissue and plasma ctDNA mutation profiles in treatment-naïve ovarian cancer and investigated its clinical implications. Our findings demonstrate that tissue–plasma concordance varies substantially among patients and is significantly associated with clinicopathological factors, tumor molecular characteristics, and clinical outcomes. These results provide novel insights into the utility and limitations of liquid biopsy in ovarian cancer management.
The overall concordance rate of 40.8% in our cohort is comparable to those reported in other advanced solid tumors, including studies demonstrating rates of 50.0% in gastric cancer, 24 46.0% in non-small-cell lung cancer, 25 and 39.6% in prostate cancer, 26 though direct comparisons are challenging due to variations in methodology and gene panels. This moderate concordance rate reflects both technical and biological factors. Technically, we found that mutations detected exclusively in tissue samples had significantly lower VAF compared to shared mutations, suggesting that detection sensitivity in plasma might be a limiting factor. However, the correlation between tissue and plasma VAFs for shared mutations suggests that, despite technical limitations, plasma ctDNA analysis can modestly reflect the relative abundance of genomic alterations when successfully detected. Biologically, the higher proportion of functionally relevant mutations among shared variants indicates that driver mutations are more likely to be detected in plasma, potentially due to their higher abundance in tumor cells and selective pressure maintaining them at detectable levels.28,29
The lower concordance rates for SVs compared to SNVs, particularly for CNVs among SV subtypes, highlight the technical and biological challenges of detecting complex genomic alterations in plasma ctDNA. This is especially relevant in ovarian cancer, where genomic instability characterized by frequent CNVs is a hallmark feature. 30 Detecting SVs in cfDNA requires a high tumor fraction for accurate analysis. Previous studies have employed detection thresholds of 15%–25%,30,31 which substantially exceeds the median plasma ctDNA fraction observed in our cohort, potentially leading to underestimation of variant prevalence. These findings underscore the need for enhanced sequencing methodologies and bioinformatic algorithms to improve SVs detection in genomic instability cancers. 31 Recently, liquid biopsy by low-coverage whole-genome sequencing (0.1–0.5×) has emerged as a suitable method to quantify chromosomal instability in ovarian cancer. 32 In addition, a CNV calling pipeline developed by Nguyen et al. 33 was able to detect high copy-count amplifications in plasma samples with tumor fractions below 1%.
Precision medicine requires genomic profiling to guide targeted therapy, traditionally achieved through invasive tissue sampling. In ovarian cancer, this poses particular challenges due to peritoneal dissemination, spatial heterogeneity, and technical difficulties in obtaining adequate samples. Liquid biopsy through ctDNA analysis has emerged as a promising minimally invasive alternative, with approved assays now available and demonstrated clinical utility in gynecologic malignancies. 11 However, our findings reveal important limitations to universal replacement of tissue biopsy. While ctDNA detected additional mutations not identified in tissue samples (potentially addressing spatial heterogeneity), 57.5% of patients showed poor concordance (<50%), with ctDNA failing to detect substantial tissue-identified mutations. Therefore, the critical need is to precisely select patients suitable for ctDNA genomic profiling by identifying factors that influence tissue–plasma concordance.
To our knowledge, this is one of the first studies to comprehensively explore the influence of clinicopathological characteristics on tissue–plasma genomic concordance through inter-individual analysis in ovarian cancer. Our findings demonstrated that tissue TMB and plasma ctDNA fraction were independently associated with tissue–plasma concordance, which is supported by several previous reports in other cancer types.26,34 Advanced tumor stage is also a significant predictor of high concordance according to previous studies, 35 as also evidenced by the predominance of early-stage disease in the poorly concordant group in our study. However, this association was not confirmed in our subsequent logistic analyses, possibly due to the limited number of early-stage patients. Notably, studies in other solid tumors have demonstrated higher tissue–plasma concordance rates when tumor tissue is obtained from metastatic sites rather than primary lesions, with distinct variations also observed across different metastatic locations, including liver, lung, and peritoneum.20,36 Given that ovarian cancer frequently presents with advanced-stage disease and extensive peritoneal dissemination, we investigated the relationship between tumor sampling location and tissue–plasma concordance, as well as the impact of visceral metastasis status. However, no significant association was observed. This finding is consistent with a previous study in ovarian cancer, 28 indicating that the tumor mutational evolution during metastasis in ovarian cancer is different from other malignancies.
Although our study showed no significant differences in BRCA or HRD status between concordance groups, ctDNA-tissue concordance patterns may still relate to HRD-associated biological processes. Mechanistically, deficient DNA damage repair pathways convert single-strand breaks to lethal double-strand breaks, leading to cancer cell death and subsequent ctDNA release.37,38 In HRD-positive tumors, this process creates a paradox: while increased ctDNA shedding enhances plasma detectability, the genomic instability generates large SVs such as chromosomal rearrangements and CNVs, 39 which pose significant technical challenges for detection, as our study demonstrates. This dual effect may explain the lack of significant BRCA or HRD status differences between concordance groups in our study. Therefore, distinct ctDNA detection strategies may be needed for different HRD statuses to optimize detection accuracy. For HRD-positive tumors, specialized algorithms for SVs detection may be necessary.
The association between concordance rate and clinical outcomes in our study is intriguing. Patients with poor tissue–plasma concordance demonstrated reduced chemosensitivity as evidenced by lower KELIM scores. This reduced chemosensitivity translated to poorer clinical outcomes, with the poorly concordant group exhibiting elevated early recurrence risk, which was confirmed by analysis of 12-month PFS—a critical prognosis indicator and timepoint for defining complete platinum sensitivity in ovarian cancer.40,41 These findings suggest that tissue–plasma concordance might potentially serve as a prognostic marker reflecting tumor biology and treatment sensitivity beyond the technical aspects of ctDNA detection. However, the small sample size and limited follow-up duration necessitate cautious interpretation of these results. The prognostic value of tissue–plasma concordance has been demonstrated in other solid tumors.24,25,42 Several potential biological mechanisms may underlie this association. First, low concordance could reflect greater intratumoral heterogeneity, a characteristic consistently linked to unfavorable treatment responses and survival outcomes across diverse cancers. 43 Second, discordant genomic profiles might signal active and aggressive clonal evolution within the tumor, potentially driving the development of treatment resistance. 4 Third, the pattern of ctDNA release into circulation likely reflects fundamental tumor characteristics, including the extent of necrosis and apoptosis, degree of angiogenesis, and regulation of ctDNA-containing extracellular vesicle secretion, which may collectively influence treatment response. 44
While limited concordance rates in treatment-resistant patients may raise questions about ctDNA reliability, it is essential to distinguish between different aspects of ctDNA prognostic value. Previous studies demonstrating the prognostic significance of ctDNA detection status itself (positive vs negative during treatment monitoring, reflecting tumor burden below the detection threshold of conventional examinations) remain valid, 12 as this prognostic value stems from detection status rather than concordance with tissue genomic profiles. Our study expands this utility by identifying baseline tissue–plasma concordance patterns as an additional prognostic marker, complementing rather than contradicting existing findings. Future studies should investigate how baseline tissue–plasma concordance patterns influence the interpretation and prognostic value of subsequent ctDNA monitoring during treatment.
Our study is strengthened by the prospective design with standardized sample collection and treatment protocols implemented by a single medical team, minimizing operational variations. All tissue and plasma samples were collected simultaneously prior to treatment initiation, allowing evaluation of fundamental concordance at baseline, without confounding effects of systemic therapy and temporal sampling variations documented in previous studies.45,46 However, certain limitations should be acknowledged. First, the sample size is relatively small with an underrepresentation of early-stage patients, potentially limiting generalizability. In addition, inherent technical biases from our sequencing methodology may have influenced the results. Most importantly, our ctDNA sequencing panel omitted BRCA1/2, substantially limiting clinical relevance given that BRCA1/2 mutations are among the most actionable alterations in ovarian cancer. Second, follow-up time is short, which may affect the long-term prognostic assessments. Third, our concordance cutoff was based on studies in other tumor types due to the absence of ovarian cancer-specific data. Future validation studies incorporating larger cohorts, comprehensive BRCA1/2 testing, and ovarian cancer-specific concordance thresholds are warranted.
Conclusion
In conclusion, our prospective study systematically evaluated the genomic concordance between tumor tissue and plasma ctDNA in ovarian cancer and explored its clinical significance. We identified key predictors of concordance and revealed their novel association with treatment responses and survival outcomes. These findings provide important insights for optimizing liquid biopsy implementation in clinical practice, though larger studies are needed for further validation.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251399468 – Supplemental material for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications
Supplemental material, sj-docx-1-tam-10.1177_17588359251399468 for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications by Hao Su, Rong Fan, Mingle Tian, Yuan Li, Yongxue Wang, Tao Wang, Sha Wang, Xirun Wan and Fengzhi Feng in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359251399468 – Supplemental material for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications
Supplemental material, sj-docx-2-tam-10.1177_17588359251399468 for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications by Hao Su, Rong Fan, Mingle Tian, Yuan Li, Yongxue Wang, Tao Wang, Sha Wang, Xirun Wan and Fengzhi Feng in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-3-tam-10.1177_17588359251399468 – Supplemental material for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications
Supplemental material, sj-docx-3-tam-10.1177_17588359251399468 for Pretreatment circulating tumor DNA and tissue genomic profiling concordance in ovarian cancer: a prospective observational study of influencing factors and clinical implications by Hao Su, Rong Fan, Mingle Tian, Yuan Li, Yongxue Wang, Tao Wang, Sha Wang, Xirun Wan and Fengzhi Feng in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We would like to thank patients and their families for participating in the study. We would like to thank the investigators and their support staff who generously participated in this work.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.