
Abstract
Background:
Circulating tumor DNA (ctDNA) analysis that is tumor-informed and personalized requires high-quality tissue specimens, which are unavailable in certain clinical contexts and pathology practice in Southeast Asia.
Objectives:
We aimed to develop and clinically validate an alternative tumor-naïve ctDNA assay.
Design:
A retrospective observational study.
Methods:
Our tumor-naïve multimodal profiling integrated mutation detection, using both amplicon and hybridization sequencing, with analysis of copy number alteration (CNA) and fragmentomics of cfDNA. We analyzed blood samples of 948 cancer patients and 566 non-cancer donors enrolled in previous studies to evaluate the analytical performance of ctDNA detection. Clinical performance was assessed using post-surgical samples of 97 breast cancer and 51 colorectal cancer patients to compare tumor-naïve ctDNA status with clinical recurrence. The performance was directly compared with the tumor-informed method using identical samples.
Results:
For mutations, a combination of amplicon and hybridization sequencing provided higher sensitivity and broader coverage of mutation detection than single methods. Variants of clonal hematopoiesis of intermediate potential were common, mainly in the TP53 gene, and must be excluded. Besides mutations, the addition of CNA and fragment length profiles significantly improved the sensitivity of ctDNA detection in the metastatic stage, but modestly in the early stage. In breast cancer, surveillance tumor-naïve ctDNA achieved 54.5% sensitivity and 98.8% specificity for predicting recurrence (hazard ratio (HR) = 23.3, p < 0.0001). In colorectal cancer, the sensitivity and specificity of surveillance ctDNA for predicting recurrence were 80.0% and 100%, respectively (HR = 35.6, p < 0.0001). The overall accuracy of the tumor-naïve method was lower than the tumor-informed method, but the performance gap varied by cancer stage and cancer type.
Conclusion:
The tumor-naïve method could be a reliable alternative to monitor ctDNA when obtaining high-quality tissue samples is challenging. The performance of this method was better in high ctDNA-shedding cancer or at the metastatic stage.
Keywords
Introduction
Circulating tumor DNA (ctDNA) has been demonstrated as a noninvasive biomarker to detect minimal residual disease (MRD), predict recurrence, and monitor treatment response in multiple solid tumors.1–3 Currently, there are two approaches for ctDNA-MRD testing: tumor-informed and tumor-naïve methods. The tumor-informed approach customized for patient-specific mutations is generally more sensitive and specific to detect ctDNA than the tumor-naïve approach.4,5 However, it necessitates high-quality tissue samples, which are challenging when tissue biopsy is unobtainable in certain clinical contexts, or when the specimens have suboptimal quality, a common issue in developing countries. 3
The tumor-naïve approach not only overcomes issues of tissue availability and heterogeneity but also offers the advantages of rapid turnaround time and real-time monitoring of tumor evolution. This approach is not personalized but utilizes pre-designed panels to detect certain genetic and epigenetic features characteristic of ctDNA. Mutations alone are usually not sufficient, and integration of other non-mutation features such as methylation, fragment length, and copy number variations has demonstrated significant improvement in ctDNA detection sensitivity.6,7 Several studies have reported promising performance of the tumor-naïve approach in tumors with high ctDNA-shedding rate, such as early-stage colorectal cancer 6 and metastatic lung cancer 8 ; but the performance was lower in tumors with low ctDNA-shedding, like breast cancer.7,9 Despite substantial evidence from various patient cohorts, there is a notable lack of research investigating the reliability and accuracy of tumor-naïve ctDNA-MRD testing in Southeast Asia.
In this retrospective study, we validated a tumor-naïve multimodal profiling assay that integrates mutation, copy number variation, and fragmentomics to detect ctDNA in Vietnamese patients with different types of cancer. Both technical and clinical performance of the assay were evaluated, and then directly compared with our previously established tumor-informed assay using an identical set of samples and sequencing platform.
Materials and methods
Patients and sample collection
This retrospective study utilized existing samples and clinical information of Vietnamese patients enrolled in our previous clinical trials and a real-world study assessing tumor-informed ctDNA monitoring in multiple solid tumors. The study design, eligibility criteria, and sample collection have been published for the prospective clinical trials of colorectal cancer,10,11 breast cancer,12,13 and the real-world multi-cancer study. 3 In total, pre-treatment blood samples of 948 patients and 566 non-cancer donors were included to assess the technical performance of ctDNA detection using the tumor-naïve method. For clinical performance to predict recurrence in early-stage cancer, post-surgical plasma samples of 97 breast cancer patients (plasma = 220) and 51 colorectal cancer patients (plasma = 92) were analyzed to compare the ctDNA status with pre-existing records of clinical recurrence. Patient demographics are listed in Table S1. The reporting of this study conforms to the STROBE statement (Supplemental File 1).
Tumor-informed personalized ctDNA assay
Formalin-fixed paraffin-embedded (FFPE) tissue and matched white blood cell (WBC) samples were already sequenced for 155 cancer-associated genes (Table S2). Plasma samples were already processed and analyzed for ctDNA status using personalized tumor-specific mutations in our previous studies.3,10,12,13
Tumor-naïve multimodal ctDNA assay
To detect mutations in the plasma, cfDNA libraries barcoded with unique molecular identifiers were prepared by the xGen™ cfDNA Library Prep v2 MC kit (Integrated DNA Technology, Coralville, Iowa, USA) according to the manufacturer’s protocol. Libraries were pooled and hybridized with custom probes targeting 22 genes (Integrated DNA Technology; Table S2) and then sequenced with an average depth of 500×. Multiplex PCR (mPCR) was performed in a separate reaction to amplify approximately 500 hotspot mutations from cfDNA, followed by ultra-deep amplicon sequencing with an average depth of 100,000×. To analyze genome-wide non-mutation features, part of the cfDNA libraries prepared above were subjected to shallow whole-genome sequencing (sWGS) at the average depth of 0.5×.
Variant calling from both hybridization capture and amplicon sequencing was described previously.10–13 For all variants found positive in cfDNA, we used gDNA of WBC to amplify those positions by mPCR and sequenced at 10,000× to exclude germline variants 14 and variants indicative of clonal hematopoiesis of intermediate potential (CHIP). CHIP variants were determined by variant allele frequency (VAF) in WBC at 0.1%–10% based on a previous study 15 ; some were further confirmed as having unchanged VAF in the longitudinal blood samples of the same patients.
sWGS data were then used to determine fragment length profile (FLEN) and end-motif (EM) signatures as previously described. 16 Briefly, the 9th column in the BAM files corresponding to the DNA fragment length values was extracted. Fragment lengths ranging from 50 to 350 bp were selected to construct the FLEN features, resulting in a 301-dimensional feature vector. For 256 EMs, we calculated the total absolute difference between the motif frequency of a cancer sample and the average motif frequency in non-cancer samples. Copy number alteration (CNA) and CNA-based tumor fraction (TF) estimates were obtained using the ichorCNA workflow. 17 The dataset was then partitioned into train (cancer = 400, non-cancer = 240), test (cancer = 100, non-cancer = 60), and independent validation (cancer = 264, non-cancer = 266) subsets. The train and test sets were used to transform FLEN vectors into NMF_FLEN values using non-negative matrix factorization as previously described. 18 The NMF_FLEN, EM score, and ichorCNA values were used as classification scores of cancer and non-cancer. Thresholds for classification were then determined for each feature in the train set and applied to the test and validation sets to calculate the area under the curve (AUC) of the receiver operating characteristic curves, as well as to assess the sensitivity and specificity to distinguish cancer and non-cancer samples.
For TF determination, if mutations were detected, the sample TF was the mean VAF of all positive mutations. When mutations were absent, sample TF, equal to the non-mutation TF, was the ichorCNA value, if positive, or the converted NMF_FLEN signal using an in-house linear regression algorithm.
Statistical analysis
The correlation between non-mutation TF and mutation VAF was examined using Pearson correlation analysis. The comparison of ctDNA detection rates among different methods was performed by Chi-square and Fisher’s exact tests. Kaplan–Meier estimation was used to generate survival curves, and group differences were assessed by the log-rank test. Cox proportional hazard regression was employed to calculate the hazard ratio (HR). All statistical analyses were performed in GraphPad Prism, with significance defined as p < 0.05.
Results
Study design
Our tumor-informed approach sequenced paired FFPE and WBC for 155 genes to select top personalized mutations, which were amplified by mPCR followed by ultra-deep sequencing to detect plasma ctDNA (Figure 1). When high-quality tumor tissue is not available or insufficient, our tumor-naive approach utilizes pre-designed panels to identify ctDNA. For mutations, hybridization capture using a 22-gene panel and mPCR using a 500-hotspot panel were used simultaneously. For non-mutation features, sWGS was used to profile CNA, fragment length, and end-motif profiles of cfDNA (Figure 1).

Workflows of tumor-informed and tumor-naïve ctDNA analysis. Tumor-informed approach requires high-quality FFPE samples that are sequenced for 155 genes to identify tumor-specific mutations; ctDNA was then detected by top-ranked personalized mutations. Tumor-naïve approach does not require FFPE and utilizes pre-designed panels of both mutation and non-mutation features to identify ctDNA. For mutations, ctDNA was detected by both hybridization capture using a 22-gene panel and multiplex PCR using a panel of 500 hotspot mutations. For non-mutation features, shallow WGS and a machine learning model were used to detect ctDNA.
The study utilized plasma samples of 948 cancer patients (458 early stage I–III and 490 metastatic stage IV patients) and 566 non-cancer donors (Figure 2(a)). For analytical validation of mutation detection, mPCR and hybridization capture workflows were compared for the type, the breadth, and the depth of variants being detected. For non-mutation features, the train, test, and validation datasets were used to optimize the classification thresholds of single features and select the best feature combination to identify ctDNA. The final tumor-naïve method was then assessed for clinical performance of pre-treatment ctDNA detection and cancer recurrence prediction (Figure 2(a)).

Study design and technical performance of the tumor-naïve method. (a) Plasma samples of 948 cancer patients and 566 non-cancer donors were used. Samples were first used for analytical validation of mutation detection by mPCR and hybridization capture using fixed panels, and non-mutation feature detection using machine learning models, with the data being split into train, test, and independent validation sets. Clinical performance was then assessed for pre-treatment ctDNA detection and cancer recurrence prediction. The number of patients and donors in each analysis was shown. (b) For mutations, the number and VAF of all mutations detected by mPCR and hybridization capture methods were compared. (c) Frequency of the most common genes that had CHIP mutations in different cancer types. (d) VAF of CHIP mutations found in the matching WBC and cfDNA samples. (e) ctDNA detection performance using single genomic features was measured by the ROC curves and associated AUC values. (f) Performance of different feature combinations. (g) High correlation of non-mutation TF with mutation VAF values (p < 0.05, Pearson correlation), and distribution of sample TF levels among cancer and non-cancer samples.
Analytical performance to detect tumor-naïve ctDNA
In 501 treatment-naïve cancer samples, a total of 565 mutations were detected in 382 samples, of which mutations detected by both mPCR and hybridization accounted for 33.8% (191/565; Figure 2(b)). Mutations detected solely by mPCR accounted for 55.8% (315/565), and had much lower VAF (median 0.24%) than the shared mutations (median 7.19%). The hybridization workflow identified an additional 10.4% (59/565) mutations, mostly at high VAF (median 7.00%), and they were either SNPs and indels not covered by the mPCR panel, or fusion variants undetectable by amplicon sequencing (Figure 2(b)). In addition, CHIP filtering by WBC sequencing was found to be critical as CHIP variants were present in all cancer types, most commonly in the TP53 gene (Figure 2(c)). VAF of CHIP mutations in WBC had a broad range (0.10%–7.81%) that was similar to VAF in the matching cfDNA samples (0.11%–11.13%; Figure 2(d)).
For non-mutation features, the single-feature model using NMF_FLEN showed the highest AUC compared to EM and CNA in all datasets (Figure 2(e)). Fragment length distribution of cancer samples was enriched with shorter cfDNA fragments than non-cancer samples (Figure S1(A)). The combination of NMF_FLEN and CNA achieved the highest sensitivity for ctDNA detection compared to the NMF_FLEN alone or other combinations in all cancer types (Figure 2(f) and Figure S1(B)). There was a high correlation between the VAF of mutations and TF of non-mutation features (Figure 2(g)). The overall sample TF could distinguish the cancer and non-cancer plasma samples in all cancer types (Figure 2(g) and Figure S1(C)).
Clinical performance to detect ctDNA and early recurrence
For the early stage, pre-treatment ctDNA detection rates using the tumor-naïve method were 51.2%, 73.3%, 30.2%, 93.2%, 50.0%, 43.4%, and 68.8% for lung, colorectal, gastro-esophageal, liver, pancreatic, breast, and ovarian cancer, respectively (Figure 3(a)). These rates were lower than the tumor-informed method by more than 10%, significantly for colorectal, gastro-esophageal, and lung cancer. The integration of non-mutation features increased the detection rate slightly by less than 10% compared to the mutation-only method for most cancers. For the metastatic stage, pre-treatment ctDNA detection rates using the tumor-naïve method were 80.0%, 82.7%, 65.9%, 85.7%, 87.5%, 66.7%, and 72.7% for lung, colorectal, gastro-esophageal, liver, pancreatic, breast, and ovarian cancer, respectively (Figure 3(b)). These were still lower than the tumor-informed method, but the difference was less pronounced than in the early stage. Non-mutation features played a bigger role in the metastatic stage as they improved the ctDNA detection rate by more than 10% compared to the mutation-only method. In the tumor-informed method, personalized mutations not covered by fixed panels accounted for more than 50% of all positive mutations, particularly for gastro-esophageal cancer (Figure 3(c) and Figure S2). A small fraction of mutations, though covered by the fixed panel, remained undetected by the hybridization method of the tumor-naïve approach (Figure S2). These explain its overall lower sensitivity compared to the tumor-informed approach.

Pre-treatment ctDNA detection rate in different cancer types and stages. Comparison of ctDNA detection rates among the tumor-naïve methods using mutation only, using both mutation and non-mutation features, and the tumor-informed method using personalized mutations, in the (a) early stage I–III and (b) metastatic stage IV. (c) The percentage of shared and unique mutations detected by the tumor-naïve method, using mPCR and hybridization capture, versus the tumor-informed method.
To evaluate the performance of tumor-naïve ctDNA to predict recurrence, plasma samples at the landmark time point of 1 month after surgery and at follow-up visits were retrospectively analyzed for early-stage breast (n = 97, plasma = 220) and colorectal (n = 51, plasma = 92) cancer patients (Figure 4(a)). For breast cancer, disease-free survival (DFS) of patients stratified by tumor-naïve surveillance ctDNA, not the landmark ctDNA, was significantly different. Patients with ctDNA(+) had a significantly higher risk of recurrence than those with ctDNA(−) results (HR = 23.3, p < 0.0001; Figure 4(b)). The 24-month DFS of patients with ctDNA(+) was 28.6% while the DFS of those with ctDNA(−) was 95.4%. Sensitivity and specificity of surveillance ctDNA to predict recurrence were 54.5% and 98.8%, respectively, with the mean lead time of 5.5 months (Figure 4(b)). This performance of the tumor-naïve method was much lower than that of the tumor-informed method, which achieved the sensitivity, specificity, and mean lead time of 90.9%, 98.8%, and 8.8 months, respectively. Non-mutation features did not improve performance for the tumor-naïve method (Figure 4(b)). For colorectal cancer, the DFS of patients stratified by both tumor-naïve landmark and surveillance ctDNA status was significantly different (HR = 13.3 and 35.6, respectively, p < 0.0001). The 24-month DFS of patients with surveillance ctDNA(+) was 0% while the DFS of those with ctDNA(−) was 94.3% (Figure 4(c)). Sensitivity and specificity of surveillance ctDNA to predict recurrence were 80.0% and 100%, respectively, with the mean lead time of 5.7 months (Figure 4(c)). This performance was still lower than that of the tumor-informed method that achieved the sensitivity, specificity, and mean lead time of 90.0%, 100%, and 7.1 months, respectively. The addition of non-mutation features improved the sensitivity of recurrence detection by 10% compared to the mutation-only method. Swimmer plots depicting longitudinal ctDNA status and clinical outcome of each relapsed case are illustrated in Figure S3.

Performance of tumor-naïve ctDNA to predict recurrence in early-stage cancer. (a) Two cohorts of early-stage breast and colorectal cancer patients were included. Blood samples at 1 month after surgery (landmark) and at follow-up visits were analyzed for ctDNA status. For (b) breast cancer and (c) colorectal cancer, Kaplan–Meier analysis of disease-free survival for patients stratified by tumor-naïve ctDNA status either at the landmark time point or during surveillance (all post-operative time points). Sensitivity, specificity, and mean lead time of surveillance ctDNA to predict recurrence were compared among the tumor-naïve method, using mPCR and hybridization capture, and the tumor-informed method.
Finally, we presented case studies to highlight the significance of all three components of the tumor-naïve method (Figure 5). For the early stage, patient ZMB016, a 41-year-old female diagnosed with stage III, luminal B breast cancer, had ctDNA detected 9.7 months before clinical recurrence. There was a clear correlation of TF level and postoperative ctDNA status determined by either mutations by mPCR or by hybridization, as well as non-mutation features. Patient ZMC006, a 65-year-old male patient with stage IIA colon cancer, also had ctDNA detected 3.5 months earlier than clinical recurrence. Mutations by mPCR were the most sensitive to detect low levels of ctDNA in this case (Figure 5(a)). For the metastatic stage, patient ZMB111, a 36-year-old female patient with metastatic breast cancer, had ctDNA persistence after chemotherapy, correlating with the later confirmation of progressive disease. Only non-mutation features of ctDNA were detected in this case. Lastly, patient ZML112, a 68-year-old female patient with stage IIIB lung cancer harboring an EGFR Del19 mutation, was treated with Afatinib. ctDNA was persistently positive during treatment, and the patient eventually progressed. All features of ctDNA were detected and well-correlated in this case; a new resistance mutation, EGFR T790M, was identified, which helped to inform subsequent therapeutic intervention (Figure 5(b)).
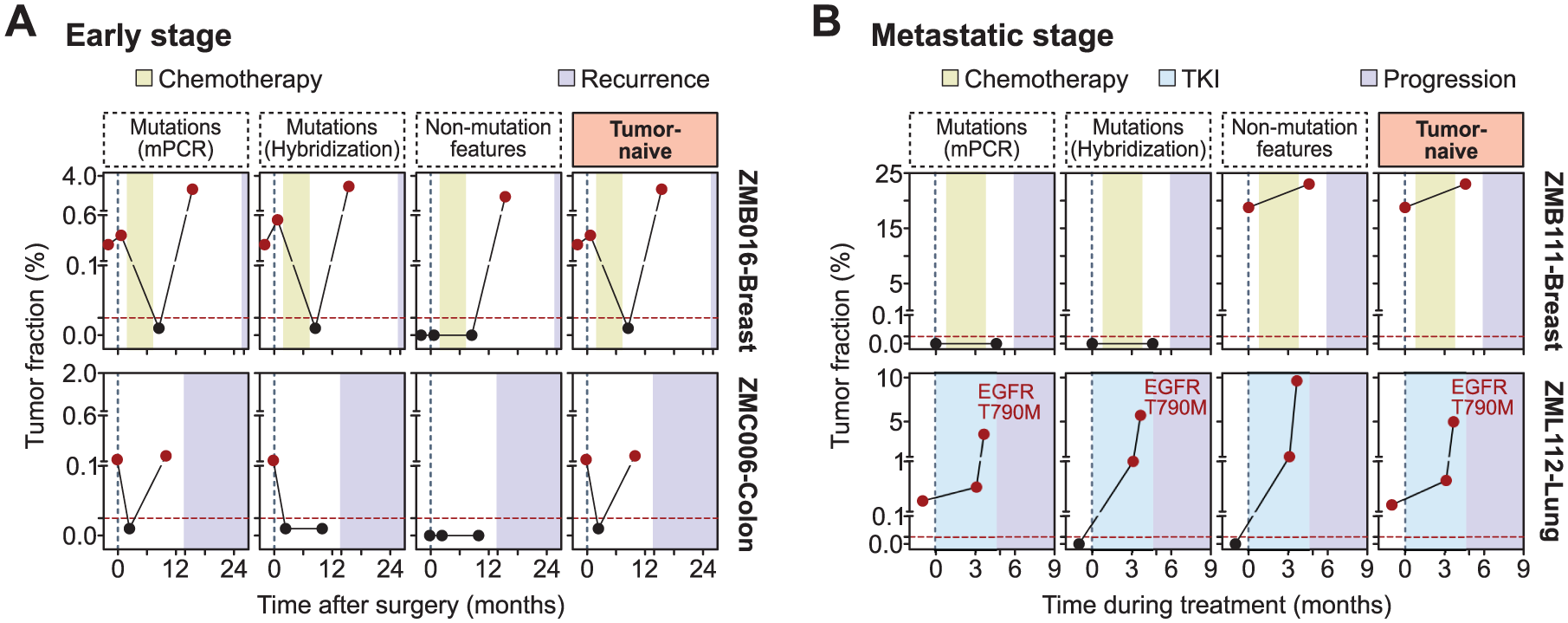
Case studies of tumor-naïve ctDNA detection. (a) For early-stage cancer, tumor-naïve ctDNA could detect residual cancer before clinical recurrence. Case ZMB016 demonstrated the strong correlation of TF levels and ctDNA status determined by all three components of the tumor-naïve method: mutations by mPCR or hybridization, and non-mutation features by machine learning modeling. Case ZMC006 illustrated the high sensitivity of the mPCR method to detect low levels of ctDNA. (b) For the metastatic stage, the dynamics of ctDNA levels could predict disease progression before clinical diagnosis. Case ZMB111 highlighted the important role of non-mutation features for ctDNA monitoring. Case ZML112 demonstrated the high correlation of TF levels and ctDNA status determined by all three components and emphasized the capability to identify resistance mutations of the tumor-naïve method.
Discussion
In this study, our tumor-naïve method combined both mutation and non-mutation features to improve sensitivity for ctDNA detection. For mutations, we leveraged the advantages of both workflows: the high depth, high sensitivity, and high specificity of targeted amplicon sequencing, and the broad mutation coverage of hybridization capture sequencing, especially for fusion variant detection in lung cancer. CHIP filtering was found critical for the tumor-naïve method, as we observed CHIP mutations most frequently in the TP53 gene and at VAF below 1%, similar to other reports. 15 For non-mutation features, the majority of studies used epigenetic alterations to identify ctDNA-MRD (Table S3). While the ctDNA methylation profile is a sensitive and specific marker, the workflow to process samples using bisulfite or enzyme conversion for methylation analysis is labor- and cost-intensive. In our wetlab protocol, we utilized part of the cfDNA libraries already prepared for mutation hybridization to run sWGS for genome-wide feature analysis, so that the process was more streamlined and cost-effective. In recent studies, the features CNA and fragmentomics have been demonstrated to correlate with tumor burden and VAF of mutations,19,20 which agrees with our findings.
Our tumor-naïve method combining mutation, CNA, and fragment length features achieved higher accuracy to detect ctDNA and recurrence than several tumor-naïve assays using either mutation or methylation alone (Table S3). When using both mutation and methylation, Nakamura et al.’s 21 study reported 81.0% sensitivity and 98% specificity to predict recurrence in colorectal cancer, which was comparable with our performance. For recurrence prediction in breast cancer, Janni et al.’s studies, also combining mutation and methylation, reported either lower sensitivity at 28.9% 9 or similar sensitivity at 54.5% 7 in comparison with our study. This finding suggests that combining mutation with genome-wide features other than methylation holds significant potential to monitor ctDNA-MRD.
The overall performance of our tumor-naïve ctDNA analysis was lower than the tumor-informed approach. This has been the observation across independent studies using these two approaches separately in different cancer types. 22 However, very few have performed a head-to-head comparison of these methods using the same set of samples, same DNA input, same reagents, and sequencing platform like our study. A recent cross-platform evaluation of five commercial ctDNA assays revealed that the mutation detection of these assays was unreliable at VAF below 0.5%, especially with low DNA input. 23 Therefore, our study provided the unique direct comparison and indeed confirmed that the tumor-informed method tracking personalized mutations by mPCR resulted in better sensitivity and specificity for ctDNA detection, agreeing with Santonja et al. 5 that also directly compared the two methods in breast cancer.
The first reason for the lower performance of the tumor-naïve method was that more than 50% of ctDNA mutations were patient-specific and not covered by fixed panels. Such distinctiveness in the mutation profiles of cancer patients has also been documented. 24 Although designing a larger fixed panel would help improve coverage, it should be noted that many mutations in cancer-associated genes could be benign or linked with old age rather than cancer in different individuals. 25 Therefore, to avoid false positivity, our panel design primarily targets driver or hotspot mutations that are more likely to be linked with cancer. Second, we found that for early-stage cancer when tumor burden is low, the gap in performance between the tumor-informed and tumor-naïve methods was greater, and the role of non-mutation features was minimal. This indicates that the sensitivity of our tumor-naïve method must be improved, using machine learning models integrating multiple genome-wide features of ctDNA, or WGS data at a depth higher than 1X, to better detect the early-stage MRD. In metastatic cancer, the performance difference between the two methods is smaller, likely because of the high tumor burden requiring a lower limit of detection. 26 Non-mutation features significantly improved the ctDNA detection rate, probably due to higher genomic abnormalities in the tumor genome at this stage. 27 Third, we observed higher sensitivity of the tumor-naïve method in lung, colorectal, and liver cancers, but lower sensitivity in breast and gastro-esophageal cancers. This indicates that the performance of this method also varied by cancer types, depending on their ctDNA-shedding rate, mutation load, and involvement of genome-wide alterations.
Limitations of our study include the small sample size for each cancer type and the retrospective design, which analyzes existing samples. Future prospective clinical trials, particularly interventional ctDNA-guided studies, are essential to fully evaluate the performance of our assay and to demonstrate the clinical utility of ctDNA-MRD monitoring in clinical practice. Moreover, further optimization of non-mutation features is also required to enhance the sensitivity of ctDNA detection, and machine learning models to recognize CHIP variants in cfDNA could also be explored to complement or replace WBC sequencing.28,29 Despite these shortcomings, our study demonstrated the feasibility and reliability of the tumor-naïve method when obtaining high-quality tissue samples is challenging.
Conclusion
In summary, we propose that the tumor-informed method should be the gold standard for MRD detection in the early stage, especially for low ctDNA-shedding tumors like breast and gastro-esophageal cancers. The tumor-naïve method could be used to detect MRD in high ctDNA-shedding cancers like colorectal cancer, provided sufficient clinical validation of the test. Both methods are reliable for monitoring ctDNA in different cancer types at the metastatic stage.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251393090 – Supplemental material for Tumor-naïve multimodal profiling of circulating tumor DNA to detect minimal residual disease in solid tumors
Supplemental material, sj-docx-1-tam-10.1177_17588359251393090 for Tumor-naïve multimodal profiling of circulating tumor DNA to detect minimal residual disease in solid tumors by Tu Nguyen, Van-Anh Nguyen Hoang, Trong Hieu Nguyen, Trung Hieu Tran, Ngoc Nguyen, Tho Thi Le Vo, Duy Sinh Nguyen, Hoa Giang, Hoai-Nghia Nguyen and Lan N. Tu in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.