
Abstract
Epithelial ovarian cancer (OC) comprises molecularly distinct disease types, with high-grade serous ovarian cancer (HGSOC) accounting for ~75% of OC diagnoses; ovarian clear cell carcinoma (OCCC) and endometrioid ovarian carcinoma (EnOC) at ~10% each; mucinous ovarian carcinoma (MOC) and low-grade serous ovarian carcinoma (LGSOC) at ⩽5% each; and ovarian carcinosarcoma (OCS), the rarest type of OC at 1%–4% of OC diagnoses. LGSOC has the best prognosis, followed by EnOC, MOC and OCCC, with HGSOC then OCS being the most aggressive. For all types of OC, diagnosis at the advanced-stage results in dramatically reduced survival. Initial treatment consists of debulking surgery and platinum-based chemotherapy, usually in combination with a taxane; however, response rates vary depending on the OC type. Treatments specific to the OC type may improve treatment outcomes. For HGSOC, poly(ADP-ribose) polymerase inhibitor (PARPi) therapy has improved survival for women with DNA homologous recombination repair (HRR) defects; however, acquired resistance remains an issue and more effective treatments are needed. Next-generation sequencing of distinct types of OC has revealed the complexity of genetic variants and larger-scale genomic and epigenomic alterations harboured, including proven and putative biomarkers of drug response. A predominance of distinct gene classes is altered in specific OC types: HRR genes (e.g. BRCA1 and BRCA2) in HGSOC; ARID1A and PIK3CA in OCCC; PIK3CA and KRAS in EnOC; CDKN2A and KRAS in MOC and MAPK pathway genes (e.g. BRAF and KRAS) in LGSOC. Generating evidence for effective drug combination therapies targeting relevant aberrations in each OC type is urgently needed. The effects of long-term drug treatment on OC genomes, acquired drug-resistance and OC relapse require clarification, especially in women with HGSOC with acquired resistance to PARPi. This article provides an overview of the main types of OC and their genomic profiles. It highlights recent encouraging clinical trials, with an emphasis on the future of genomically-targeted combination therapies, for both first-line and subsequent treatment of OC. We focus on PARPi combinations for HGSOC, MAPK pathway inhibitors for LGSOC, cell cycle checkpoint inhibitors for OC with CCNE1 amplification, the potential of immune checkpoint inhibitors in OCCC and encouraging, as yet preliminary, responses for antibody-drug conjugate-based therapy. Thus, OC type-specific genomic susceptibilities provide direction for personalised therapy in OC.
Plain language summary
Ovarian cancer (OC) can be divided into distinct disease types based on the cell type from which the cancer originated, with high-grade serous ovarian cancer being the most prevalent OC type. Each type of OC can be further classified by changes in the DNA which affect specific genes. Treatment options are increasing as a result of the number of drugs in clinical trials shown to have efficacy in matching these DNA changes. Changes in the DNA may affect cancer cell growth and survival, DNA repair, or interaction of the cancer cells with the immune system. Finding effective drug combinations to target these changes is essential, to most effectively treat patients. In addition, understanding how long-term drug treatment may lead to drug resistance and OC relapse is crucial. In this review, we describe the most frequent DNA or protein changes observed in the different types of OC and discuss different therapies currently being investigated to target these changes. We focus on PARP inhibitors, MAPK pathway inhibitors, cell cycle checkpoint inhibitors, immune checkpoint inhibitors, and antibody-drug conjugate (ADC)-based therapies for specific OC types depending on genomic susceptibilities.
Keywords
Background of epithelial ovarian cancer
In 2020, over 300,000 individuals were diagnosed with ovarian malignancies worldwide and over 200,000 died as a result of their disease. 1 This article is confined to the consideration of epithelial OC, which comprises six main types that can be distinguished from one another by their histologic and molecular profiles. 2 We took a non-biased approach in our literature selection, using combinations of terms, such as ‘ovarian cancer’, ‘prognosis’, ‘standard treatment’ and ‘molecular profiling’, as well as searching https://clinicaltrials.gov/ for any trials involving ovarian cancer (OC) and targeted therapies. Putative cells of origin may occur in the ovary (including endometriotic cysts), fallopian tube or in endometrial tissue (from endometriosis/endometrium). 2 In addition, predominantly high-grade serous (HGS) malignancies may be defined as being either of ovarian (high-grade serous ovarian cancer, HGSOC), fallopian tube (HGSFTC) or primary peritoneal carcinomas (PPC), based on the predominant site of disease at diagnosis, and are treated similarly due to their related behaviour and outcomes. 3 Whilst the histological classification is currently considered when deciding on treatment strategies, the molecular profile should also be taken into account to optimise outcomes. Here, we will discuss the potential of precision medicine in OC, which matches relevant targeted therapies to pathogenic variants (PV), either germline or somatic, of genes that alter regulators of normal cellular functions, such as proliferation, apoptosis, cell cycle and DNA repair, to give a patient the best chance of survival.
Pathological subtypes and incidence
HGSOC is the most prevalent type of OC, comprising ~75% of total OC diagnoses and has been shown to derive from the distal fallopian tube secretory epithelial cells.4,5 Next, both ovarian clear cell carcinoma (OCCC) and endometrioid ovarian carcinoma (EnOC) comprise ~10% of diagnoses each. 4 These types of OC derive from endometriotic cells, which may be present in endometriotic cysts on the ovarian surface, or within areas of pelvic endometriosis, initially deriving from the endometrial lining of the uterus. 4
Mucinous ovarian carcinoma (MOC) and low-grade serous ovarian carcinoma (LGSOC) each comprise ⩽5% of diagnoses. 4 Analysis of mucinous borderline ovarian tumours (MBT) and associated low- and high-grade MOC in specific patients supports a progressive model of carcinogenesis for MOC. 6 Therefore, MOC likely derives from ovarian surface epithelium, with some suggestion of derivation from ovarian teratoma.6,7 LGSOC, on the other hand, may derive from the fallopian tube epithelium, similar to HGSOC. 8 Finally, ovarian carcinosarcoma (OCS) is the rarest type of OC at 1%–4% of diagnoses, and is composed of a biphasic population of carcinomatous and sarcomatous cells, likely undergoing epithelial-to-mesenchymal transition (EMT).9,10 Molecular analyses carried out by us and others have indicated that OCS are monoclonal, likely deriving from HGSOC or EnOC.9–15 We also recently demonstrated that OCS most likely arises by the conversion theory, as carcinomatous components isolated from OCS samples had higher EMT gene scores than purely HGSOC samples. 9 This reveals how molecular subtleties can help distinguish OC disease states.
Survival rates
The survival rate of patients with OC depends on the type and stage of OC at diagnosis, 16 and as is becoming increasingly appreciated, the molecular profile of each individual tumour. Serous borderline tumours and MBT both have an excellent prognosis with 5-year survival (5-YS) rates of 95% and 97%, respectively. 17 However, MOC have a lower overall 5-YS rate of 63%–71%.16,17 Dramatically, the 5-YS rate drops to <20% for those diagnosed with MOC at an advanced stage.4,16,17 Genomic profiling of disease stages for the different OC types in recent years has uncovered putative drivers of disease progression. For example, MOC likely evolves from MBT, with enrichment of TP53 PVs as well as copy number aberrations (CNAs) during progression. 6 EnOC have overall 5-YS rates of 69%–82% and, similar to MOC, the presence of TP53 PVs and increased CNAs are associated with advanced disease and a worse prognosis.16–18 In contrast, EnOC with CTNNB1 PVs were more likely to be diagnosed at an early stage and had a better prognosis. 19 These studies highlight the importance of careful disease staging, as well as molecular profiling, in choosing the most appropriate treatment strategies.
For OCCC, the overall 5-YS rate of 54%–66% drops to 10% for those diagnosed at a late stage.16,17 LGSOC has a 5-YS rate of 85% for those who achieved complete cytoreduction, but only 32% for individuals with residual disease (RD). 20 For HGSOC, the 5-YS rate is 43% overall, but just 9% for those diagnosed with stage IV disease.16,17 Finally, OCS has the poorest overall 5-YS rate of just 21%, due to most diagnoses occurring at later stages of disease and the more pluripotent nature of this biphasic disease, with a greater potential to undergo EMT and transform into a more aggressive disease type.16,17
Current standard treatment strategies
For all types of OC, initial management consists of primary debulking surgery (PDS) and adjuvant platinum-based chemotherapy, usually in combination with a taxane.21,22 In this setting, surgery plays an important role in achieving no RD, which is the most important prognostic factor in OC. 23 In cases of unresectable disease or the presence of comorbidities, neoadjuvant chemotherapy (NACT) and interval debulking surgery are considered, although better overall survival (OS) is observed with PDS. 24 Bevacizumab, a monoclonal antibody targeting vascular endothelial growth factor (VEGF), has since been included in treatment protocols with first-line chemotherapy and also as maintenance therapy. 25 More recently, poly(ADP-ribose) polymerase (PARP) inhibitors (PARPi) have been added as first-line maintenance therapies for individuals with homologous recombination-deficient (HRD) tumours, and in some cases, irrespective of homologous recombination repair (HRR) status. 26 The most responsive type of OC to standard treatment is HGSOC, with an objective response rate (ORR) of ~80%. 27 The response rate of other OC types to chemotherapy is significantly lower, particularly for OCCC (ORR ~30%), which is largely considered to be resistant to current standard chemotherapy agents. 4 This highlights the need for OC type-specific treatments to improve treatment outcomes for these patients. The advent of PARPi therapies for the treatment of HRD OC, predominantly for HGSOC/HGSFTC/PPC, has increased the life expectancy of some patients, but acquired resistance to PARPi over time remains a roadblock to treatment efficacy. 28 Therefore, the interest in targeting alternative aberrations is a major focus of OC research and clinical trials.
Genomic landscape
As described above, each type of OC represents a distinct disease. This is reflected in the genomic profile of each OC type, with an overview provided in Tables 1–3. For HGSOC, the most common genetic aberrations occur in the tumour suppressor gene TP53, with 90%–98% of patients having somatic TP53 PVs.29–33 HGSOC are usually genomically unstable, with ~50% being HRD. 34 This is due to specific molecular features, such as the presence of PVs in key HRR genes, such as BRCA1 or BRCA2, or high-level (homozygous) promoter methylation of either BRCA1 or RAD51C.34–37 For example, germline BRCA1/BRCA2 PVs occur in 13%–22% of HGSOC patients, with somatic PVs in 5%–8% of HGSOC.29,30,32,34,38–40 Germline or somatic loss-of-function of other HRR tumour suppressor genes, such as RAD51C, RAD51D, PALB2 and BRIP1, have also been reported in ~6% of HGSOC patients.38,39 Other genes commonly inactivated in HGSOC include NF1 (5%–17%), RB1 (2%–11%) and PTEN (1%–6%).29,33,41 The most commonly amplified genes in HGSOC include CCNE1 (7%–23%), MECOM (~27%), MYC (9%–34%) and PIK3CA (3%–22%).29,32,40,41
Commonly altered genes in ovarian cancer.
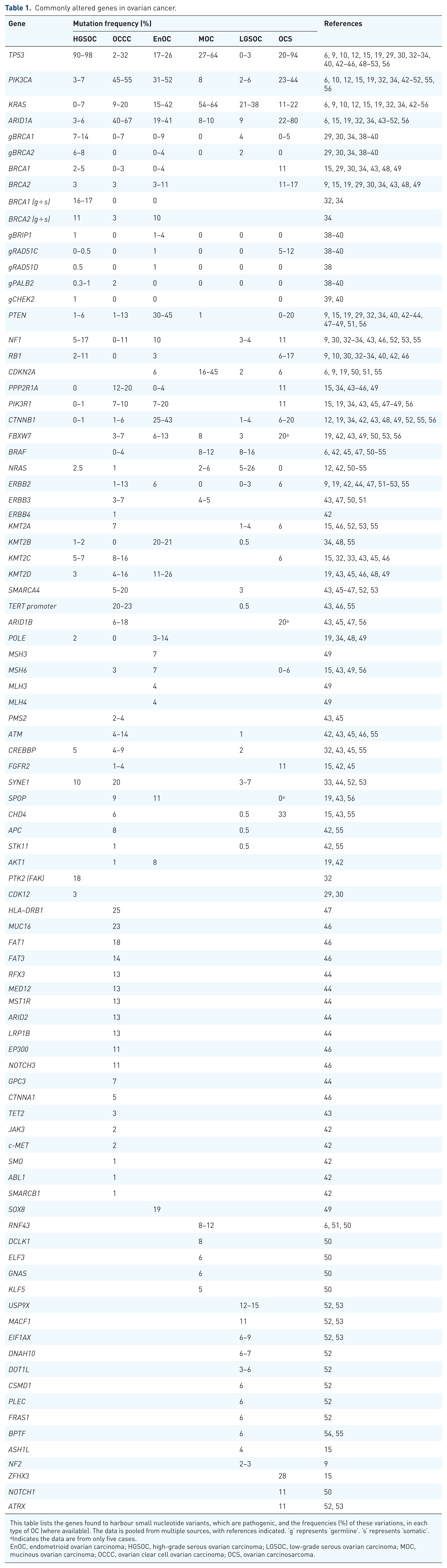
This table lists the genes found to harbour small nucleotide variants, which are pathogenic, and the frequencies (%) of these variations, in each type of OC (where available). The data is pooled from multiple sources, with references indicated. ‘g’ represents ‘germline’. ‘s’ represents ‘somatic’.
Indicates the data are from only five cases.
EnOC, endometrioid ovarian carcinoma; HGSOC, high-grade serous ovarian carcinoma; LGSOC, low-grade serous ovarian carcinoma; MOC, mucinous ovarian carcinoma; OCCC, ovarian clear cell ovarian carcinoma; OCS, ovarian carcinosarcoma.
Commonly amplified genes in ovarian cancer.

This table lists the genes found to be amplified, and the frequencies (%) of these amplifications, in each type of OC (where available). The data is pooled from multiple sources, with references indicated.
EnOC, endometrioid ovarian carcinoma; HGSOC, high-grade serous ovarian carcinoma; LGSOC, low-grade serous ovarian carcinoma; MOC, mucinous ovarian carcinoma; OCCC, ovarian clear cell ovarian carcinoma; OCS, ovarian carcinosarcoma.
Commonly deleted genes in ovarian cancer.

This table lists the genes found to be deleted, and the frequencies (%) of these deletions, in each type of OC (where available). The data are pooled from multiple sources, with references indicated.
EnOC, endometrioid ovarian carcinoma; HGSOC, high-grade serous ovarian carcinoma; LGSOC, low-grade serous ovarian carcinoma; MOC, mucinous ovarian carcinoma; OCCC, ovarian clear cell ovarian carcinoma; OCS, ovarian carcinosarcoma.
In contrast to HGSOC, OCCCs have a relatively low frequency of PVs in TP53 (2%–32%) and have very low frequencies of BRCA1/BRCA2 PVs.34,38,42–46 OCCCs commonly have PVs in ARID1A (40%–67%), PIK3CA (35%–55%), the TERT promoter (20%–23%), PPP2R1A (12%–20%) and KRAS (9%–20%).34,42–47 Other low-frequency events, which are still of interest for potential targeted therapies, include PVs in the oncogenes BRAF, ERBB2/3 and 4, AKT1 and PTEN.42,43,45 The most commonly amplified genes in OCCC are GNAS (~47%), MYC (~40%), NTRK1 (~33%), AKT2 (9%–25%), ERBB2 (9%–11%) and ZNF217 (7%–62%).44,47 In addition, deletions of CDKN2A/B and ARID1A are found in 3%–20% and 29% of OCCCs, respectively.34,44 In EnOCs, TP53 PVs are present in 17%–26% of cases and the BRCA1/BRCA2 PV rate is ~3%–15%, usually in the high-grade TP53-mutant EnOC.34,38,48 The genes most commonly altered are CTNNB1 (25%–43%), PIK3CA (31%–52%), KRAS (15%–42%), ARID1A (19%–41%) and PTEN (30%–45%).19,34,48,49 Less common PVs occur in POLE (3%–14%) and mismatch repair (MMR) genes (5%–19%), which lead to MMR deficiency (MMRd).19,34,48,49
In MOC, CDKN2A is lost (either through PV or copy number loss) in a large proportion of cancers (up to 76%) and PVs occur in TP53 (27%–64%), KRAS (54%–64%), BRAF, PIK3CA, ARID1A and RNF43 (~8%–12% for each).6,50,51 ERBB2 amplification occurs in 16%–27% of MOCs and HRD and MMRd are both very rare.6,50,51
In LGSOC, the MAPK pathway genes, KRAS (21%–38%), BRAF (10%–18%) and NRAS (5%–26%), are most commonly altered and PVs in the tumour suppressors NF1 and NF2 occur in 2%–4% of cases each.52–55 Other potential driver variants have been identified in USP9X (12%–15%), MACF1 (11%), ARID1A (9%) and EIF1AX (6%–9%).52,53 In addition, LGSOC can harbour homozygous deletion of CDKN2A/2B (1%–12%) and USP9X (1%–10%).52,53,55
Finally, OCS most commonly harbour TP53 PVs (20%–94% patients).9,10,12,15,56 PVs in PI3K-pathway genes (e.g. PTEN, PIK3CA and PIK3R1) are also found in >50% of patients as well as frequent PVs in ARID1A (>20%), KRAS (11%–22%), BRCA2 (11%–17%), CTNNB1 (6%–20%) and RB1 (6%–17%).9,10,12,15,56 Amplifications of MYC, MECOM and CCNE1 are also frequently seen in OCS.9,15
Targeted therapies for the treatment of OC
Despite the number of genetic variants and their known effects in epithelial OC, therapies to target many of these pathogenic events in cancer cells have not been developed or progressed to the clinic, as yet. In addition, there are currently no FDA-approved drugs for OC that target the effects of TP53 inactivation, KRAS constitutive activation, CDKN2A loss, AKT2 amplification, MYC amplification or PIK3CA constitutive activation. This is disappointing as these aberrations are present in one or more types of OC, as described above. Indeed, when the National Cancer Institute-Molecular Analysis for Therapy Choice (NCI-MATCH) phase II trial (NCT02465060) applied a next-generation sequencing (NGS) panel to sequence samples from 75 OC patients, only 14% of these patients had an actionable genetic alteration, highlighting the need for better targeted therapies. 57 Panels have evolved since then to include a broader range of targets, matched to a wider array of therapies. However, NGS panels have evolved since then to include a broader range of targets matched to a wider array of therapies. For example, the Australian Molecular Screening and Therapeutics (MoST) study used a larger gene panel test to sequence gynaecological cancers from 533 patients, with the goal of finding targetable variants. 58 Whilst 39% of these patients had clinically actionable variants, only 3.5% accessed treatment, further highlighting the need for more targeted treatments to be available at a suitable time in the disease journey. It is also important to recognise that some genomic alterations have been well-established (mostly in other cancer types), whereas others still require further validation. For example, the response of HRD tumours to PARPi has been demonstrated in many cancer types, including OC, whereas the specific alterations in PIK3CA that will be responsive to PI3K inhibitors (PI3Ki) require further assessment. Similarly, the response of tumours with ARID1A alterations to ATR inhibitor (ATRi) therapy is becoming clearer, whereas the correlation of Programmed Death Ligand-1 (PD-L1) expression with response to immunotherapy remains ambiguous. The European Society for Medical Oncology Scale for Clinical Actionability of molecular Targets can be used to rank genomic alterations based on clinical evidence to facilitate patient selection for targeted therapies. 59 Clinical trials for individuals with OC, involving targeted therapies matched to genetic aberrations, where patient benefit was observed, are discussed below and listed in Table 4.
Clinical trials testing targeted therapies in OC.

This table lists the clinical trials that have been completed or are currently active and/or recruiting to test relevant targeted therapies in different types of OC. Only trials where clinical benefit was observed are included. Current clinical trials involving promising targeted therapies that are yet to publish results are also included.
ATRi, ATR inhibitor; CAPRI, combination ATR (ceralasertib) and PARP (olaparib) inhibitor; CBR, clinical benefit rate; CCGC, clear cell gynaecological cancer; CDH6, cadherin-6; CFI, chemotherapy-free interval; CI, confidence interval; CLDN6, Claudin-6; CR, complete response; CTLA-4, cytotoxic T-lymphocyte-associated antigen 4; D, durvalumab; Dato-DXd, Datopotamab Deruxtecan; DCR, disease control rate; DOR, duration of response; EnOC, endometrioid ovarian carcinoma; FGFR1, fibroblast growth factor receptor 1; FT, fallopian tube; gBRCAm, germline BRCA-mutated; HER2, human epidermal growth factor receptor 2; HGSOC, high-grade serous ovarian carcinoma; HR, hazard ratio; HRD, homologous recombination deficient; HRP, HRR proficient; ITT, intent-to-treat; LGSOC, low-grade serous ovarian carcinoma; MIRV, mirvetuximab soravtansine; MOC, mucinous ovarian carcinoma; MoST, Molecular Screening and Therapeutics; N/A, not available; NE, not estimable; NIVO, nivolumab; NR, not reached; OC, ovarian cancer; OCCC, ovarian clear cell ovarian carcinoma; OCS, ovarian carcinosarcoma; ORR, objective response rate; OS, overall survival; PARPi, poly(ADP-ribose) polymerase inhibitor; PCC, physician’s choice chemo; PD-1, programmed death receptor-1; PD-L1, programmed death ligand-1; PDGFR, platelet-derived growth factor; PFS, progression-free survival; PM, pembrolizumab monotherapy; PP, primary peritoneal; PR, partial response; pts, patients; R-DXd, Raludotatug deruxtecan; RUCA, rucaparib; SD, stable disease; T-DXd, Trastuzumab Deruxtecan; VEGFR1–3, vascular endothelial growth factor receptors.
Targeting DNA repair
As the most prevalent type of OC, HGSOC is the most studied, with the best defined treatment regimens to date. For the ~50% of HGSOC that lack a functional homologous recombination DNA repair pathway (HRD-positive), targeted therapy became available nearly a decade ago, with the approval of PARPi therapy, initially for BRCA1/2-mutated HGSOC. 60 The major target of PARPi is the DNA damage-sensing enzyme, PARP1, whose role in normal cells is to recruit DNA repair enzymes to sites of single-strand breaks.61,62 PARP1 may also play roles in alternative DNA repair pathways. 63 Some PARPi also inhibit other members of the PARP family, such as PARP2 and PARP3, as well as other off-target kinases. 63 PARP1 specific inhibitors (i.e. saruparib/AZD5305 and the brain-penetrant AZD9574) are being developed and are under clinical investigation. 64
PARPi efficacy in HGSOC has been transformational for some patients, with response to treatment correlating with sensitivity to platinum chemotherapy or the presence of PVs in BRCA1/2 or other HRR genes. 63 The phase III SOLO1 clinical trial (NCT01844986) tested olaparib as a single-agent first-line maintenance therapy in patients with advanced OC, with germline or somatic BRCA1/2 PVs, who had experienced a complete response (CR) or partial response (PR) with prior first-line chemotherapy treatment. Strikingly, patients treated with olaparib had a 70% lower risk of disease progression or death after a median follow-up time of 41 months, compared to patients treated with placebo maintenance therapy. 65 Five-year follow-up of SOLO1 reported that the median progression-free survival (PFS) was 56.0 months for olaparib-treated patients versus only 13.8 months for patients receiving placebo (hazard ratio (HR), 0.33; 95% confidence interval (CI), 0.25–0.43). 66 Seven-year follow-up reported OS of 67% for olaparib-treated patients, compared to 46.5% for the placebo group, an absolute survival benefit of 20.5%, although not statistically different. 67
Niraparib has also proven to be very effective for patients with advanced OC. The phase III PRIMA study (NCT02655016) assessed the efficacy of single-agent niraparib as first-line maintenance therapy in patients who had already responded to platinum-based therapy. After 24 months, OS was 84% versus 77% for the placebo group (HR, 0.70; 95% CI, 0.44–1.11). 68 After 3.5 years of follow-up, the niraparib-treated patients maintained a durable PFS compared to those in the placebo group. 69 Patients with HRD OC had a clinically significant 48% reduction in the risk of progressive disease (PD) or death. 69 Of interest, a clinically significant 35% reduction in the risk of PD or death was reported for the HRR proficient (HRP, or HRD-negative) group. 69 Therefore, highlighting the efficacy of the drug in all patients, not just those with HRD, and suggesting that niraparib may act on pathogenic pathways other than HRD. Five-year follow-up for PRIMA reported a PFS in the overall population of 22% for niraparib and 12% for placebo; however, an OS advantage was not observed. 70 After 5 years, there was a striking difference in PFS results for patients with HRD-positive OC, with those in the niraparib arm twice as likely to be progression-free as those in the placebo arm (35% vs 16%, respectively). 70 The HRD-positive niraparib-treated patient population also had a prolonged median time to first subsequent therapy (TFST) compared to the placebo arm (26.9 vs 13.9 months, respectively; HR, 0.55; 95% CI, 0.43–0.71). Niraparib also prolonged the median TFST in the overall population compared to placebo (17.0 vs 12.0 months, respectively; HR, 0.74; 95% CI, 0.62–0.89). 70
In addition to the SOLO1 and PRIMA studies, the phase III ATHENA–MONO study (NCT03522246) tested the efficacy of rucaparib as single-agent first-line maintenance therapy in patients with advanced OC. The cohort comprised patients with and without BRCA1/2 PVs (i.e. both HRD and HRP patients were included), who had previously responded to first-line chemotherapy. The study concluded that the PFS survival of patients treated with rucaparib was significantly longer than those treated with placebo, and was not dependent on their BRCA1/2 or HRD status. 71 Specifically, 45.1% of the rucaparib-treated cohort were progression-free at 24 months versus only 25.4% of the placebo group. 71
Earlier PARPi studies (reviewed in Mateo et al. 72 ) included phase III clinical trials of PARPi as subsequent-line maintenance therapy in the recurrent OC setting. Impressive prolongation of PFS was demonstrated in the NOVA, 73 SOLO2 74 and ARIEL375,76 clinical trials; however, OS benefits were unclear in the recurrent setting. Indeed, although PARPi therapy remains the most promising targeted therapy for HGSOC to date, resistance to or following PARPi therapy has developed as a major challenge, even during/following first-line maintenance PARPi therapy. Finding more nuanced treatments to improve outcomes for patients has been the focus of several studies in recent years (for detailed reviews on this topic, see Bound et al., 28 Mateo et al., 72 Bhamidipati et al. 77 and Veneziani et al. 78 ).
Anti-angiogenic agents in combination with PARP inhibitor therapy
The phase III PAOLA1 trial (NCT02477644) tested first-line maintenance bevacizumab therapy, with or without olaparib. A median PFS of 22.1 months was achieved for the combination of olaparib plus bevacizumab versus 16.6 months for placebo plus bevacizumab (HR, 0.59; 95% CI, 0.49–0.72; p < 0.001). 79 In all patients with HRD-positive OC, the median PFS was 37.2 months for olaparib plus bevacizumab versus 17.7 months for placebo (HR, 0.33; 95% CI, 0.25–0.45). For patients with HRD-positive OC without BRCA PVs, the median PFS was 28.1 versus 16.6 months (HR, 0.43; 95% CI, 0.28–0.66). 79 Five-year follow-up of the HRD-positive PAOLA1 patients revealed a median PFS of 46.8 months in the olaparib plus bevacizumab arm versus 17.6 months in the placebo plus bevacizumab arm (HR, 0.41; 95% CI, 0.32–0.54), 80 demonstrating the value of the addition of olaparib to maintenance therapy with bevacizumab.
PARPi has been combined with other anti-angiogenics, such as cediranib (targets VEGF receptors 1, 2 and 3 (VEGFR1–3)), surufatinib (targets fibroblast growth factor receptor 1 (FGFR1)), apatinib (targets VEGFR2) and anlotinib (targets VEGFR, FGFR, platelet-derived growth factor and c-KIT). The proof of concept EVOLVE study assessed olaparib plus cediranib for the treatment of individuals with HGSOC following progression on PARPi alone (NCT02681237). In this heavily pre-treated cohort, there was an ORR of 0% (0/11) for platinum-sensitive patients, 20% (2/10) for platinum-resistant patients and 8% (1/13) for the exploratory cohort. 81 Another study investigated the PARPi pamiparib in combination with surufatinib (NCT05494580), also in a heavily pre-treated cohort. A median PFS of 7.5 months (95% CI, 3.4–not evaluable) was achieved, with 2 patients (11.1%) experiencing PR and 14 patients (77.8%) achieving stable disease (SD). 82 In a phase I study of the PARPi fuzuloparib plus apatinib (NCT03075462), patients with BRCA-mutated OC had an ORR of 62.5% compared to 40.9% for patients with non-BRCA-mutated OC, respectively. 83 Median PFS was 9.4 months (95% CI, 1.9–20.5 months) and 6.7 months (95% CI, 2.0–14.9 months), respectively. 83 The phase II NSGO-AVANOVA2 trial (NCT02354131) demonstrated that niraparib plus bevacizumab was more effective than niraparib alone for treating platinum-sensitive recurrent OC. 84 The median PFS for the combination was 11.9 months (95% CI, 8.5–16.76 months) compared to 5.5 months (95% CI, 3.8–6.3 months) for niraparib alone. 84 An even better response was observed in the phase II OVARIO trial (NCT03326193), which investigated niraparib with bevacizumab for patients who had responded to first-line, platinum-based chemotherapy. Median PFS was 19.6 months (95% CI, 16.5–25.1 months) for all patients, which increased to 28.3 months (95% CI, 19.9–not reached) when just considering the HRD-positive population. 85 Finally, the single-arm phase II ANNIE study (NCT04376073) investigated niraparib plus anlotinib for the treatment of platinum-resistant recurrent OC (PROC). Median PFS was 9.2 months (95% CI, 7.4–11.9 months). 86 Due to these impressive responses to PARPi plus anti-angiogenic therapy, specifically from the results of the PAOLA 1 trial, olaparib plus bevacizumab is now approved for the first-line maintenance treatment of OC.79,87
Immune checkpoint inhibitors and combinations with PARP inhibitor therapy
Re-engaging the immune system to target cancer cells that have developed mechanisms to avoid immune surveillance should be considered a promising treatment option for patients with OC. 88 To date, this has been most successful for the small proportion of OC that are MMRd and/or some OCCC. 88 Cancer cells that express PD-L1 on their surface can interact with T-cells via Programmed Death Receptor-1 (PD-1) expressed on the surface of specific subsets of T-cells, thus preventing killing of the cancer cell by the T-cell. 89 Immune checkpoint inhibitors (ICI), such as anti-PD-1 monoclonal antibodies (e.g. pembrolizumab and dostarlimab) and anti-PD-L1 monoclonal antibodies (e.g. durvalumab and atezolizumab), release this block and activate T-cell-mediated cancer cell killing. 90 ICIs are proving efficacious for patients with other types of gynaecological cancers, most notably endometrial cancer and cervical cancer (see Uterine Cancer and Cervical Cancer reviews in this Special Collection for more details). However, in OC, the efficacy of PD-1/PD-L1 inhibitors has been disappointing. For example, in the phase Ib JAVELIN study (NCT01772004), OC patients with stage III–IV advanced high-grade OCS, FTC or PPC, who were treated with the PD-L1 inhibitor (PD-L1i) avelumab, had a median PFS of just 2.6 months (95% CI, 1.4–2.8 months). 91 In addition, the phase Ib KEYNOTE-028 trial (NCT02054806) investigated the efficacy of the PD-1 inhibitor (PD-1i) pembrolizumab in treating patients with advanced metastatic PD-L1-expressing OC and found, after a median follow-up time of 15.4 months, that patients had a median PFS of just 1.9 months (95% CI, 1.8–3.5 months). 92 Clearly, ICI are unlikely to prove effective on their own in advanced OC and alternative approaches, such as combination therapies, will be required. Despite the disappointing results discussed above, some benefit of ICI monotherapy was observed in OCCC, and other advanced clear cell gynaecological cancers, in the phase II PEACOCC trial (NCT03425565). In this single-arm study, pembrolizumab demonstrated modest efficacy, with a median PFS of 12.2 weeks (95% CI, 5.9–32.9 weeks). 93 As no survival advantage was seen for patients with OCCC in the randomised MOCCA study (NCT03405454), for the PD-L1i durvalumab versus standard-of-care chemotherapy, 94 further effort is underway to identify which OCCC patients will benefit from ICI therapy.
ICI and anti-angiogenic combinations have been trialled in attempts to improve responses. For example, the combination of the anti-angiogenic agent lenvatinib with pembrolizumab demonstrated efficacy in previously treated advanced HGSOC in the LEAP-005 trial (NCT03797326). An ORR of 35% (95% CI, 19%–55%) was achieved, with a median PFS of 6.2 months (95% CI, 4.0–8.5 months). 95 Responses were observed irrespective of PD-L1 status. ICI combinations in conjunction with chemotherapy have also been trialled. The phase II TRU-D study (NCT03899610) combined durvalumab and the cytotoxic T-lymphocyte-associated antigen 4 inhibitor, tremelimumab, with NACT to treat newly diagnosed advanced OC. In this trial, a median PFS of 17.5 months (95% CI, 10.6–not reached) was achieved and the median OS was not reached. 96 The ORR of 95.7% was much improved compared to single-agent ICI studies, with 3 CR (13.0%) and 19 PR (82.6%). 96 Multiple clinical trials combining ICI with anti-angiogenics and chemotherapy have attempted to explore this further, without success to date.
The observation that exposure to PARPi resulted in increased antigen presentation on cancer cells (due to the induced DNA damage) and an increased anti-cancer immune response led to the testing of PARPi plus ICI combinations. 88 A single-arm phase II study (NCT02484404), involving treatment of a mostly (86%) PROC cohort with olaparib plus durvalumab, resulted in an ORR of 14% (5/35; 95% CI, 4.8%–30.3%) and overall median PFS of 3.9 months (95% CI, 2.8–5.9 months). 97 Another single-arm phase I/II trial in PROC (TOPACIO/KEYNOTE-162; NCT02657889) investigated the combination of niraparib with pembrolizumab, reporting an ORR of 18% (90% CI, 11%–29%) and median PFS of 3.4 months (95% CI, 2.1–5.1 months). 98 The phase III ANITA trial in platinum-sensitive relapsed (PSR) OC (NCT03598270), testing the PD-L1i atezolizumab or placebo, combined with chemotherapy and followed by niraparib maintenance, was also a negative study. 99 Finally, the combination arms of the first-line phase III ATHENA trial (ATHENA-COMBO; NCT03522246) were reported, comparing rucaparib combined with the PD-1i nivolumab, with single-agent rucaparib, in individuals with newly diagnosed, high-grade OC and found no increased benefit for the combination maintenance therapy following chemotherapy (median PFS was 15.0 months for the combination and 20.2 months for rucaparib single agent, increasing to 28.9 and 31.4 months, respectively, for HRD-positive disease). 100 These results indicated significant challenges in generating benefit from ICI therapy in HGSOC, even in first-line combination regimens.
Triple PARPi-based combinations, including ICI and anti-angiogenic therapy, have also been tested. A phase II study by the GINECO group (NCT04015739) combined olaparib with bevacizumab and durvalumab. In the PROC cohort, patients achieved a median PFS of just 4.1 months (95% CI, 3.5–5.9 months).
101
Similarly, the phase II OPAL study (NCT03574779) investigated niraparib plus bevacizumab plus the PD-1i dostarlimab in patients with PROC. The ORR was 17.1% (80% CI, 9.8–27.0) and the median PFS was 7.9 months (95% CI, 4.2–10.9 months).
102
The phase II non-randomised MEDIOLA study (
To more formally assess the combination of PARPi with anti-angiogenic therapy and ICI in the first-line OC setting, the phase III DUO-O trial (NCT03737643) investigated chemotherapy (paclitaxel/carboplatin) plus durvalumab and bevacizumab, followed by maintenance durvalumab, bevacizumab and olaparib for the treatment of newly diagnosed advanced HRD and HRP OC. Median PFS was 23.3 months for Arm 1 (chemotherapy plus bevacizumab plus placebo with maintenance bevacizumab and placebos), 25.1 months for Arm 2 (chemotherapy plus bevacizumab and durvalumab with maintenance bevacizumab, durvalumab and placebo) and 45.1 months for Arm 3 (chemotherapy plus bevacizumab plus durvalumab with maintenance bevacizumab, durvalumab and olaparib) in the HRD cohort. 105 Median PFS was 19.3 months for Arm 1, 20.6 months for Arm 2 and 25.1 months for Arm 3 in the intent-to-treat (ITT) cohort. 105 A significant improvement in PFS was observed between Arm 3 and Arm 1 for both cohorts, as well as for the HRP subgroup of the ITT cohort.105,106 However, although this suggested potential benefit for the triple combination arm, a fourth arm with olaparib maintenance therapy without durvalumab was not included, making it difficult to determine how much additional benefit was conferred by durvalumab. Therefore, it was considered premature to conclude from the three-arm DUO-O trial that all women with newly diagnosed advanced OC should be considered for complex combination chemotherapy, anti-angiogenic, PARPi and ICI therapy.
Cell cycle checkpoint inhibitors and PARPi therapy
Cell cycle checkpoint molecules, such as ATR, WEE1 and CHK1, are activated in response to DNA damage, replication stress and PARPi treatment. 107 Therefore, there has been interest in combining additional inhibitors of DNA damage response (DDRi) with PARPi for the treatment of OC. 107 This approach may also be employed for the treatment of OC that is PARPi-resistant. 108 In a phase I study (NCT03057145) of the CHK1 inhibitor (CHK1i) prexasertib with olaparib in BRCA-mutated PARPi-resistant HGSOC, four patients (22.2%) experienced PR. 109 In the phase II EFFORT trial (NCT03579316), the combination of the WEE1 inhibitor (WEE1i) adavosertib with olaparib was assessed in individuals with PARPi-resistant OC. Median PFS was 6.8 months (90% CI, 4.3–8.3 months) for the combination arm, compared to 5.5 months (90% CI, 3.9–6.9 months) for adavosertib alone. 110 In the phase II CAPRI trial (NCT03462342), the ATRi ceralasertib was combined with olaparib to treat recurrent HRD HGSOC, which had previously responded and then progressed on PARPi. The median PFS was 7.43 months (95% CI, 4.73–15.1 months). 111 In the phase II ATARI study (NCT0405269), which included OCCC and other rare gynaecological cancer types, the efficacy of ceralasertib, either alone or in combination with olaparib, was investigated in cancers with or without ARID1A loss. This was particularly relevant for OCCC, where ARID1A loss is common. 112 ATR is a serine-threonine kinase involved in single-strand DNA repair at stalled replication forks. Inhibition of ATR has been shown to be synthetically lethal in ARID1A-deficient cells, specifically in OCCC cell lines with disruptive ARID1A variants. 112 In the ATARI study, there was no association of outcome with ARID1A status observed.112,113 Median PFS was 15.4 weeks (95% CI, 7.7–23.3 weeks) for the OCCC cohort with ARID1A loss, 15.1 weeks (95% CI, 12.7–22.9 weeks) for the OCCC cohort with no ARID1A loss and 23.9 weeks (95% CI, 7.9–36 weeks) for the non-clear cell cohort. 113 To date, variability in responses of PROC and PARPi-resistant OC to DDRi combination therapies likely reflects genomic complexity, including acquired drug-resistance mechanisms, inherent in these OC, with new advances needed.
PARPi has also been tested in combination with inhibitors of other aspects of cell signalling, such as members of the PI3K/AKT/mTOR pathway. In a phase Ib study (NCT02208375), olaparib was combined with the AKT inhibitor (AKTi) capivasertib for the treatment of recurrent endometrial cancer, triple-negative breast cancer and OC. An ORR of 19% was achieved, with 83% of responders being PROC. 114 Another phase Ib study (NCT01623349) tested the combination of olaparib with the PI3Ki alpelisib in OC, with most participants having PROC. ORR of 30% and 35% were achieved for the HRD and HRP cohorts, respectively, and the median PFS was 7.2 months (95% CI, 4.9–9.0 months). 115 The different levels of effectiveness of these trials highlight the need for further work, but also that some progress is being made for some patients using combinations of targeted agents.
Cell cycle targets in cyclin E1 up-regulated OC
The most relevant type of Cyclin Dependent Kinase inhibitor (CDKi) for HGSOC is an inhibitor of CDK2 (CDK2i), as CCNE1 amplification and/or Cyclin E1 overexpression can be used as a prognostic biomarker in HGSOC. 116 CCNE1 encodes Cyclin E1, a cell cycle regulator that binds to CDK2 to mediate the transition from G1 to S phase of the cell cycle. 117 Excessive Cyclin E1 production in the absence of functional p53 disrupts the normal G1 to S transition and causes genomic instability, and is associated with a poor prognosis in OC. 117 In a recent consortium study (including results from 20 studies), the authors found that patients with CCNE1 amplification plus high protein expression had a 5-YS rate of 28.3% in comparison to a rate of 41.9% in patients with neither CCNE1 amplification nor high expression of the protein. 117 This association has been understood for some years now, but effective treatments have remained elusive. An in vitro drug-screen using HGSOC cell lines sought to discover drugs that would synergise with CDK2i to mediate the killing of CCNE1-amplified cells. 116 Drugs targeting both CDK2 and the PI3K/AKT pathway were found to be the most effective combination in CCNE1-amplified HGSOC (specifically the CDKi dinaciclib (CDK1/2/5/9 predominantly) in combination with the AKTi MK-2206). 116 These findings are yet to be translated into relevant clinical trials. More specifically, less toxic CDKi are also in development, which may be more promising in future combination therapies.
A phase II clinical trial (NCT03253679) testing the WEE1i adavosertib showed promising results in patients with CCNE1-amplified disease. 118 WEE1 allows cells with high levels of Cyclin E1 to survive by binding to CDK1/2 and preventing cell cycle progression and thus the accumulation of lethal genomic damage. Adavosertib binds to and inhibits WEE1, preventing this protective mechanism, leading to death of cells harbouring high expression of Cyclin E1. 118 In the phase II clinical trial, the CCNE1-amplified epithelial OC cohort had an ORR of 36% (95% CI, 13%–65%) and median PFS of 6.3 months (95% CI, 2.4–10.2 months). 118 In comparison, patients who did not have epithelial cancers had a lower median PFS of 2.6 months (95% CI, 2.3–2.9 months), 118 highlighting the potential this class of drug holds for patients with CCNE1-amplified OC. Similarly, patients with Cyclin E1 overexpressed PROC were treated with adavosertib in the IGNITE trial, achieving an ORR of 53% and a clinical benefit rate of 61%. 119 Although not specific for CCNE1-amplified OC, a phase II trial (NCT03414047) also assessed the CHK1i prexasertib as a single agent in PROC. There were four PROC cohorts studied in the trial, with or without BRCA-mutated disease and a range of prior therapies. An ORR of 12.1% was achieved for platinum-resistant disease, irrespective of BRCA status, and 6.9% for platinum-refractory disease. 120 Due to tolerability challenges, CDKi therapy in HGSOC will require novel, less toxic CDKi or innovative approaches to better-tolerated combination regimens.
Targeting the MAPK pathway
LGSOC patients frequently have PVs in genes encoding proteins in the MAPK pathway, such as KRAS and BRAF, leading to overactivity of the signalling pathway. 121 This, coupled with the fact that LGSOC are highly resistant to chemotherapy, has led to research into inhibitors of these proteins to provide novel therapeutic options. A phase II study (NCT00551070) assessed the efficacy of the MEK inhibitor (MEKi) selumetinib (AZD6244) in LGSOC. In this study, 63% of patients had a PFS of more than 6 months. 122 Surprisingly, response to selumetinib had no significant association with KRAS or BRAF mutational status. 122 A phase II/III trial (NCT02101788) assessing the efficacy of the MEKi trametinib in LGSOC patients also concluded promising results, with a median PFS of 13.0 months (95% CI, 9.9–15.0 months) for the trametinib-treated group compared to 7.2 months (95% CI, 5.6–9.9 months) in the standard-of-care control group. 123 Most recently, the phase III MILO study (NCT01849874) assessed the efficacy of the MEKi binimetinib in LGSOC patients. No additional benefit was observed for binimetinib treatment compared to the physician’s choice chemotherapy. 124 However, patients with MAPK pathway alterations were more likely to respond to treatment with binimetinib than patients without MAPK pathway alterations (ORR of 41% compared with 13%, respectively), 125 providing hope for the direction of this treatment option in the future.
We await further results from trials including lifirafenib, an experimental and reversible inhibitor of BRAF V600E, wild-type ARAF, BRAF, CRAF and EGFR. A phase I study (NCT02610361) was conducted on adult patients with advanced/metastatic solid tumours carrying a BRAF, NRAS or KRAS mutation. Lifirafenib demonstrated an acceptable benefit-risk profile given the safety outcomes and responses in patients with BRAF V600-mutated solid tumours, including one patient with LGSOC. 126 Lifirafenib could potentially benefit patients with MAPK pathway-associated alterations in addition to BRAF V600 mutations, including activated KRAS. A phase Ib study (NCT03905148) of lifirafenib in combination with the MEKi, mirdametinib, in patients with MAPK pathway-altered cancers is currently underway.
Targeting ERBB2/human epidermal growth factor receptor 2
ERBB2 encodes the human epidermal growth factor receptor 2 (HER2), whose normal function is to transduce signals involved in cell growth and differentiation. 127 Amplification of ERBB2 and/or overexpression of HER2 results in aggressive growth in multiple solid cancer types, including HGSOC and MOC.127,128 Inhibition of HER2 in these settings is a key area of research. Whilst amplification and 3+ overexpression of HER2 is rare in OC (1%–3%), 2+ overexpression of HER2 may occur in up to 18% of cases. 127 The effects of HER2 overexpression can be targeted, for example, by Trastuzumab Deruxtecan (T-DXd), an antibody-drug-conjugate (ADC) targeting HER2. 128 Following approval in patients with breast, gastric and non-small-cell lung cancer with HER2 overexpression, the phase II DESTINY-PanTumor02 trial (NCT04482309) investigated the effect of T-DXd in patients with other HER2-overexpressing solid cancer types, including OC. 128 An encouraging ORR was observed in the OC cohort of 45.0% (95% CI, 29.3%–61.5%), which was higher than the average ORR for all cancer types combined in the study (ORR 37.1% (95% CI, 31.3%–43.2%)). 128 Cases with 3+ expression achieved an impressive ORR of 61.3% (95% CI, 49.4%–72.4%). Patients with endometrial and cervical cancers also had impressive ORR to T-DXd (57.5% for endometrial (95% CI, 40.9%–73.0%) and 50.0% for cervical (95% CI, 33.8%–66.2%)), highlighting a promising potential role for this agent in gynaecological cancers in the future. 128 Large phase III studies are underway in OC, cervical cancer and endometrial cancer, respectively (NCT04482309 and NCT05824975 are currently recruiting). In May 2024, T-DXd received FDA approval for HER2-positive (3+) solid tumours (tumour-type agnostic approval) that had progressed after prior treatment, including OC. Many other HER2 ADCs are under investigation, including in OC. 129
Antibody-drug conjugates
Apart from the HER2-targeted ADCs, the development and investigation of ADCs targeting other tumour biomarkers has rapidly increased in recent years. Currently, there are 11 ADCs approved for the treatment of solid malignancies and more than 170 in the pipeline.129,130 Antigens such as HER2, TROP2, Folate Receptor alpha (FRα), Mesothelin, MUC16, Claudin-6 (CLDN6) and dipeptidase 3 are targets for many ADCs. 131 The first FDA-approved ADC for OC was mirvetuximab soravtansine (MIRV), which includes a payload of the maytansinoid DM4 linked to an FRα monoclonal antibody. 132 The frequency of FRα expression could be as high as 70% in primary OC and 80% in recurrent OC, indicating this is an important target in OC, particularly PROC. 133 The phase III SORAYA trial (NCT04296890) investigated the efficacy and safety of MIRV in treating patients with PROC, leading to its rapid approval. An ORR of 32.4% was achieved (95% CI, 23.6%–42.2%), with a median PFS of 4.3 months (95% CI, 3.7–5.2 months). 132 The confirmatory MIRASOL trial in FRα-positive cases was the first trial to show an OS advantage in PROC, 134 with the phase III GLORIOSA trial of MIRV with bevacizumab versus bevacizumab as maintenance in platinum-sensitive OC underway. 135 Another promising ADC yet to be approved for OC is TORL-1-23, which includes a payload of monomethyl auristatin E linked to a CLDN6 monoclonal antibody. Early data are promising in the phase I trial (NCT05103683) investigating the safety and efficacy of TORL-1-23 in advanced cancers. Six of 18 evaluable patients with OC had PR. 136 The renewed interest in ADC-based therapy has led to an increase in relevant clinical trials, with hopes that they may be a transformative approach for OC treatment.
The FDA also recently granted Breakthrough Therapy designation (BTD) to the ADC raludotatug deruxtecan (R-DXd) for patients with platinum-resistant OC, PPC or FTC. This designation applies to patients previously treated with bevacizumab who express cadherin-6 (CDH6). R-DXd is the first CDH6-directed therapy to receive BTD, a significant development given that CDH6 is expressed in approximately 65% of OC, and no CDH6-targeting therapies are currently approved. 137 This designation follows encouraging results from several ongoing clinical trials. In a phase I trial (NCT04707248), a subgroup of patients with advanced/metastatic OC who had received prior systemic therapy, treatment with R-DXd resulted in a median PFS of 8.1 months (95% CI, 4.1 months–not estimable). 138 The granting of BTD underscores the FDA’s recognition of the drug’s potential to address a substantial unmet medical need and their commitment to expediting its development and review process.
Concluding remarks
Our knowledge of the molecular features of each type of OC has advanced rapidly, giving rise to the development and testing of novel targeted therapeutics. Encouragingly, clinical trial evidence supports the efficacy of some of these agents, with the potential to broaden therapeutic choices for women whose treatment options are otherwise limited. The rationale for adding PARPi to chemotherapy and anti-angiogenic therapy in BRCA1/2-mutated or related defective OC, predominantly HGSOC, has a solid evidence base in the first-line, including the maintenance therapy context. Such combinations may provide additional benefit in PSR OC, but for PROC and PARPi-resistant OC, they lack sufficient efficacy. The study of additional DDRi, such as WEE1i in Cyclin E1-overexpressing OC, with and without PARPi or other combinations, is ongoing. MEKi has demonstrated efficacy in LGSOC, even in the absence of key MAPK pathway aberrations, indicating that alternative mechanisms of MAPK pathway disruption could be at play. However, the response to current MEKi delivered as single agents is limited. Finally, a definitive role of ICI therapy in OC has not yet been proven, with ongoing investigation in OCCC likely to be the most promising context. Indeed, studies of ICI in combination with anti-angiogenic and DNA-damaging therapies, including chemotherapy, PARPi or other DDRi, remain investigational. Successful ICI delivery may require new biomarkers, directing specific novel ICI agents, in combination. Importantly, the development of novel combination regimens will be essential for future treatment advances for the majority of patients with OC, to prevent acquired resistance to chemotherapy and PARPi. Relevant trials are ongoing, which will hopefully provide future avenues for patients, particularly in the context of drugs that are already FDA-approved for other cancer types, for example, HER2-targeted therapy in HER2 aberrant OC. MIRV is the first drug shown to provide a survival benefit in PROC expressing the target FRα, providing hope for other ADCs in this setting. Timely diagnosis and molecular analysis of OC, coupled with access to effective molecularly targeted therapeutic combinations, will be instrumental in improving patient outcomes moving forward.