
Abstract
Background:
Germline pathogenic variants (GPV) in MUTYH are rare in patients with pancreatic neuroendocrine tumors (PanNET).
Objectives:
We aimed to characterize PanNET patients with GPV and/or somatic pathogenic variants (SPV) in MUTYH.
Design:
Retrospective multicenter cohort.
Methods:
Patients with PanNET harboring MUTYH pathogenic variants (MUTYH+) were identified from centers in Brazil, Europe, and the USA. Data were extracted from medical records. Germline or somatic variants were evaluated by targeted sequencing of cancer-associated genes. The primary endpoint was to clinically characterize these patients and the secondary endpoint was overall survival (OS). Aggressive behavior was defined as rapid progression, transformation to neuroendocrine carcinoma (NEC)-like histology and/or OS of <2 years from first-line treatment. All cases were diagnosed by expert pathologists.
Results:
In total, 23 patients with MUTYH+-associated PanNET were identified (17 germline; 4 somatic). Median age was 48 years (22–72), 65% were male, 12 (52%) had first-degree family history of cancer, and 70% had synchronic metastatic disease. Tumors were of G1, G2, and G3 in 17%, 52%, and 31%, of cases, respectively, and median KI-67 was 12% (2%–80%). Seven of 11 (64%) G1/G2 PanNET with tumor biopsy upon progression evolved to G3 (3 NEC-like). Median OS of 19 patients with metastases was 4.6 years (95% confidence interval (CI): 2.4–6.8), with 12 (63%) cases demonstrating aggressive behavior. MUTYH p.Gly396Asp and p.Tyr179Cys were the most frequent variants (mostly GPV). Of 13 tumors arising in patients with germline MUTYH mutations and tested for loss of heterozygosis of the wild allele, 6 (50%) exhibited a second hit event in MUTYH, with 4 demonstrating aggressive behavior. Tumor mutation burden (TMB) was low in the six treatment-naïve PanNET (TMB: 0–9) with this information, but three of seven tumors molecularly profiled after alkylating drugs and/or radioligand therapy had high TMB (TMB: 97, 65, and 728).
Conclusion:
MUTYH mutation-associated PanNET (germline and/or somatic) is associated with aggressive and high-grade disease when metastatic.
Introduction
Pancreatic neuroendocrine tumors (PanNET) are rare neoplasms associated with heterogeneous symptoms, behavior, and evolution. When metastatic, median overall survival (OS) varies significantly, depending on several factors, including tumor grade. For instance, patients with grade 1 or 2 metastatic PanNET have a median OS of 5 years, 1 while those with grade 3 PanNET, typically survive for a median of 2–3 years. 2 Yet, even within the tumor grade subgroups, heterogeneous outcomes are observed, encouraging the investigation of further prognostic biomarkers.
Molecular profiling studies in PanNET have identified somatic pathogenic variants (SPV) in genes involved in four main pathways: chromatin remodeling, DNA damage repair, activation of mammalian target of rapamycin (mTOR) signaling, and telomere maintenance.3,4 ATRX/DAXX and MEN1, genes involved in chromatin remodeling, are the most common somatic mutations (40%–50% of sporadic PanNET), followed by alterations in genes involved in the mTOR pathway (14%–25%).3,5 Additionally, whole genome sequencing has revealed that up to 17% of patients with PanNET also harbor germline pathogenic variants (GPV), including newly identified variants in DNA repair genes MUTYH, CHEK2, and BRCA2.4–6 However, given their rarity, the clinical implications of such GPV have not been fully evaluated. SPV in MUTYH appear to be relatively rare, but anecdotally, there is a perception of higher aggressiveness among cases of MUTYH mutated advanced PanNET (SPV and/or GPV).
MUTYH is a base excision repair gene, involved in the protection of DNA from mutations caused by oxidative stress. Germline biallelic inactivation of MUTYH causes the autosomal recessive MUTYH-associated colorectal polyposis syndrome and is associated with somatic G:C > T:A transversions in the APC gene, the driver of colorectal polyps and adenocarcinoma. 7 There is also an increased risk of duodenal, ovarian, and bladder cancers in biallelic carriers, 8 while monoallelic GPV carriers may have increased risks for gastric, hepatobiliary, and endometrial cancer.9,10 Recently, it has been demonstrated that MUTYH deficiency caused by GPV also occurs in PanNET, 4 but how this affects the biological behavior of these tumors remains undetermined.
Here, we have undertaken a multicenter and retrospective cohort of patients with PanNET and GPV and/or SPV in MUTYH with the objective to evaluate their clinical characteristics and outcomes.
Methods
Patients with histologically proven diagnosis of well-differentiated PanNET of any grade and stage, and a GPV or SPV in MUTYH (MUTYH+) were retrospectively identified from institutional databases of centers in Brazil, Europe, and USA. All cases were classified by pathologists who are experts in the diagnosis of neuroendocrine neoplasms (NEN). The reasons underlying the identification of MUTYH variants were: (1) consecutive unselected cases (AC Camargo Cancer Center, Haukeland University Hospital, Memorial Sloan Kettering Cancer Center, and University of California), meaning that all patients with a diagnosis of PanNET were enrolled in research projects of germline testing, regardless of family or personal history of cancer; or (2) patients with aggressive PaNET, where the attending physician ordered an NGS somatic panel to guide treatment decisions on targeted therapies (Moffitt Cancer Center and one case from AC Camargo Cancer Center). Neuroendocrine carcinomas (NEC) at diagnosis were excluded. Most commonly, GPV were identified from screening of the consecutive and unselected cases with neuroendocrine tumors (e.g.,), and SPV were identified through tumor comprehensive genomic profiling of aggressive cases (Table 1).
Overview of MUTYH GPV and/or SPV in PanNET cases with multigene germline testing and/or tumor comprehensive genomic profiling.

ACC, AC Camargo Cancer Center, São Paulo, Brazil. HUS, Haukeland University Hospital, Bergen, Norway. MSK, Memorial Sloan Kettering Cancer Center, New York City, USA. Moffitt, Moffitt Cancer Center, Tampa, USA. UCSF, University of California San Francisco, San Francisco, USA. VR, University and Hospital Trust of Verona, Italy.
GPV, germline pathogenic variants; NEN, neuroendocrine neoplasm; PanNET, pancreatic neuroendocrine tumor; SPV, somatic pathogenic variants; VAF, variant allele frequency.
Because we were interested in evaluating the clinical course in patients with MUTYH+ PanNET, we classified the clinical behavior of each case as either aggressive or indolent. A case was deemed aggressive if it was rapidly progressive (at diagnosis or during the disease course), required treatment with chemotherapy, had NEC-like behavior, or survival of less than 2 years from diagnosis of metastatic disease. These criteria were agreed upon by all of the authors, all with clinical experience in the treatment of NET patients.
The following clinical characteristics and treatment-related outcomes were extracted from medical records: age and tumor stage at diagnosis, sex, race, tumor grade (by the World Health Organization 2019 classification) 11 and histological differentiation at diagnosis, and upon progression on cancer-directed treatments (if this information was available), tumor functionality, family and personal history of cancer, performance status, sites of metastases and tracer uptake on somatostatin-receptor imaging and/or 18F-FDG-PET, type of treatments, response and duration of response, dates of therapies, and date of death or last follow-up.
Multigene germline testing and tumor comprehensive genomic profiling were done in the context of research or standard of care using a variety of commercial or institutional platforms. All panels included genetic sequencing of MUTYH. Detailed information on MUTYH analyzes and tumor biomarkers were gathered, if available. For GPV and SPV carriers, we collected data on reference transcripts, variant allele frequency (VAF) in germline and somatic testing, microsatellite instability status, and tumor mutational burden (TMB) status. Occurrence of double hit in MUTYH was considered when the VAF indicated loss of heterozygosity (LOH) of the wild-type allele in the tumors (VAF >65%). 12
Statistics and endpoints
Our primary endpoint was to clinically characterize MUTYH+ (SPV and/or GPV) PanNET cases. The secondary endpoint was OS from first-line systemic therapy. Descriptive statistics were used to summarize the population characteristics. OS was estimated by the Kaplan–Meier method and calculated from the date of diagnosis of metastatic disease (date of biopsy or radiological evidence of metastases, whichever came first) until death from any cause. The follow-up time was estimated by the Reverse Kaplan–Meier method. All analyses were performed using the STATA version 17, StataCorp LLC.
Ethical aspects
Each institution had ethical approval for their cases and/or chart review.
The EQUATOR guideline was followed to report our results.
Results
Clinical characteristics
Considering the centers who performed germline testing for consecutive patients, 12 (2.9%) out of 402 PanNET patients harbored a MUTYH GPV and one, a SPV. Ten additional selected cases were identified by somatic next-generation sequencing tests which were performed for clinical reasons or availability of research projects (Table 1).
Overall, our population is composed of 23 cases, with 17 patients presenting a PanNET in the setting of a confirmed (N = 14) of very likely (N = 3; VAF in the tumor >65%) MUTYH GPV.
Median age of MUTYH+ PanNET patients was 48 years, 65% were male, 8% had a personal history of cancer, 65% had family members with cancer, and 70% of patients were metastatic at diagnosis (Table 2). Seven patients were diagnosed with localized PanNET: one patient presented metastatic disease after 17 months from surgical resection of the primary tumor, and another, 24 months after the initial diagnosis; 5 (22%) patients remain recurrence-free at a median follow-up of 20 months.
Baseline characteristics of patients.

Two patients with G3 NET were considered ambiguous morphology G3 NET-NEC (#7 MCC and #8 MCC on Table 3).
From 16 G1/2 NET, 9 became clinically aggressive (rapidly progressive). Of these, 8 had tumor biopsy upon progression. Seven cases remained indolent and did not perform another tumor biopsy. One patient with initial G2 NET showed an aggressive progression of disease but did not undergo a biopsy to investigate if the tumor evolved to G3 NET.
Liver and/or bone metastases could be present at diagnosis or emerge during disease course.
ACTH, adrenocorticotropic hormone; PanNET, pancreatic neuroendocrine tumor; VIP, vasoactive intestinal peptide.
Nearly one-third (31%) had initial grade 3 NET (two of them with ambiguous G3 NET/NEC histology) and 56% of initially grade 1 or 2 tumors became clinically aggressive NET. Of 11 patients with grade 1 or 2 initial PanNET who had tumor biopsy performed upon progression, 7 (64%) evolved to G3 (3 NEC-like).
Twenty-six percent of patients had functioning PanNET (including 13% non-functional at diagnosis which became functioning). Two patients had gastrinomas at diagnosis and one had an ACTHoma at diagnosis; the other cases developed hormone-secretion upon tumor progression: one had a non-functioning G3 (KI-67 25%) PanNET which became an insulinoma after progression on first-line lanreotide; one patient had a non-functioning G2 (KI-67 5%) PanNET which became a VIPoma after progression on second-line everolimus, and one G2 PanNET became a gastrin and glucagon-secreting G3 NET (ambiguous histology NEC-like features) after progression on second-line GEMOX, 18 months after the initial diagnosis.
Genomic alterations
Genomic characterization of 13 of the 23 patients showed that the MUTYH p.Gly396Asp pathogenic variant was the most frequent (30%; seven patients, with four cases with a GPV only, and two with both GVP and SPV), followed by p.Tyr179Cys (four patients, all GPV). Among six patients tested for both GPV and SPV, three presented both variants. Three additional cases were considered to harbor a double hit event in MUTYH in their PanNET because of a VAF >65%. Eighteen of 23 patients had tumors evaluated for microsatellite stability, with all being MSS.
Thirteen patients had tumors molecularly profiled either before any systemic treatment (N = 5) or upon progression on alkylating agents and/or a peptide-receptor radionuclide therapy (PRRT; N = 8). Overall, the most common alterations were found in genes associated with chromatin remodeling (ATRX, DAXX, ARID1A), histone methyltransferases (MEN1), and mTOR pathway (PTEN, TSC2). TMB varied from zero to more than 700 mutations per megabase, depending on the timing of NGS: treatment-naïve PanNET harbored exclusively low TMB, while hypermutation was exclusively identified in tumors profiled after treatment with alkylating agents and/or PRRT, and presented defects in mismatch repair genes (MLH1, MSH2, MSH6; Table 3).
Genomic and clinical characterization of patients with PanNET and GPV and/or SPV in MUTYH.
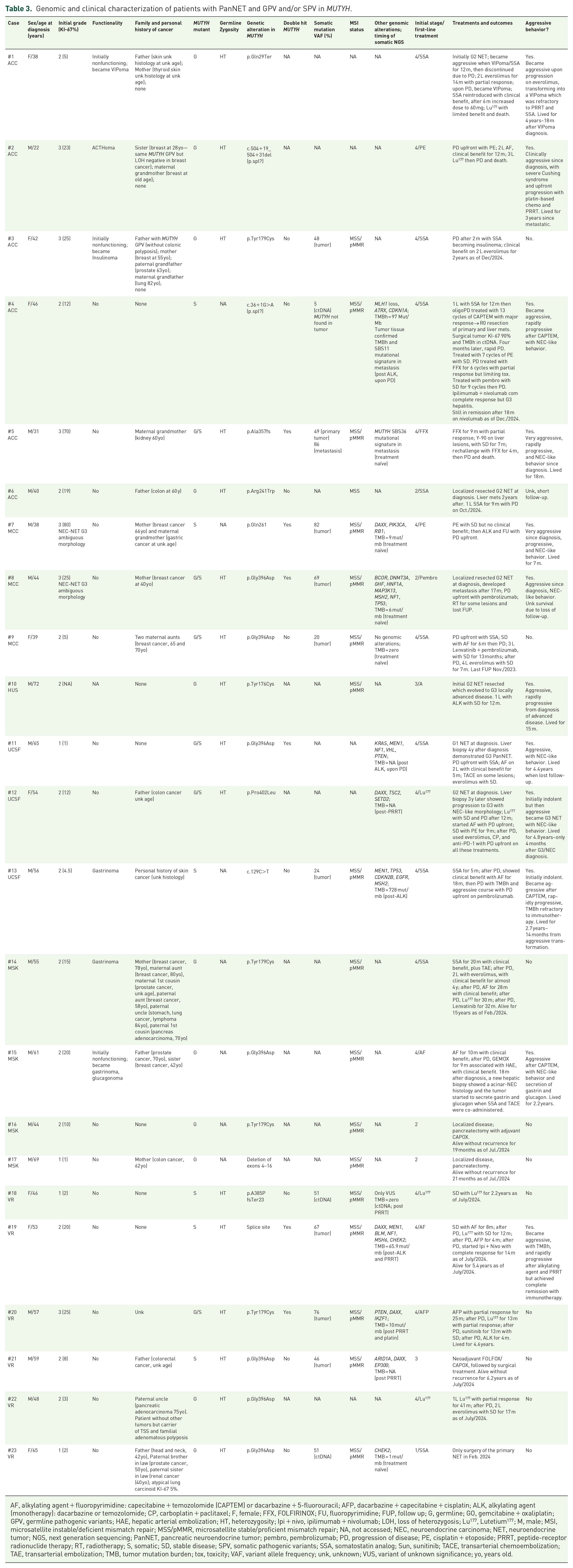
AF, alkylating agent + fluoropyrimidine: capecitabine + temozolomide (CAPTEM) or dacarbazine + 5-fluorouracil; AFP, dacarbazine + capecitabine + cisplatin; ALK, alkylating agent (monotherapy): dacarbazine or temozolomide; CP, carboplatin + paclitaxel; F, female; FFX, FOLFIRINOX; FU, fluoropyrimidine; FUP, follow up; G, germline; GO, gemcitabine + oxaliplatin; GPV, germline pathogenic variants; HAE, hepatic arterial embolization; HT, heterozygosity; Ipi + nivo, ipilimumab + nivolumab; LOH, loss of heterozygosis; Lu177, Lutetium177; M, male; MSI, microsatellite instable/deficient mismatch repair; MSS/pMMR, microsatellite stable/proficient mismatch repair; NA, not accessed; NEC, neuroendocrine carcinoma; NET, neuroendocrine tumor; NGS, next generation sequencing; PanNET, pancreatic neuroendocrine tumor; pembro, pembrolizumab; PD, progression of disease; PE, cisplatin + etoposide; PRRT, peptide-receptor radionuclide therapy; RT, radiotherapy; S, somatic; SD, stable disease; SPV, somatic pathogenic variants; SSA, somatostatin analog; Sun, sunitinib; TACE, transarterial chemoembolization; TAE, transarterial embolization; TMB, tumor mutation burden; tox, toxicity; VAF, variant allele frequency; unk, unknown; VUS, variant of unknown significance; yo, years old.
Clinical outcomes
Seven patients presented with initial localized disease, two had a later recurrence. Nineteen patients had metastatic disease. The median follow-up of all cases was 11.6 years. Systemic treatments varied and included somatostatin analogs, cisplatin-based chemotherapy regimens, alkylating-based therapies, mTOR inhibitor, and PRRT. Somatostatin analogs were the most frequent initial treatment (44%), followed by alkylating-based therapies (CAPTEM or dacarbazine plus 5-fluorouracil; 17%).
Median OS from first-line treatment of 19 metastatic patients was 4.6 years (95% CI: 2.4–6.8). Among these 19 metastatic patients, 12 (63%) exhibited aggressive evolution of their PanNET, either at diagnosis (N = 5) or upon progression (N = 7) with G3/NEC-like behavior (Table 3). Of six patients who harbored a second hit event in MUTYH in their tumors, five presented aggressive disease: three at diagnosis and one, upon progression on alkylating drug. One patient with a SPV with VAF of 76% had an indolent PanNET. One patient with LOH presented the characteristic MUTYH SBS36 signature (defined by an excess of G:C → T:A transversions). He was a 31-year-old male with a nonfunctioning aggressive G2 PanNET at diagnosis (initial KI-67 was 12% in hepatic bulky metastases) and with a GPV (p.Ala357fs). He initially had tumor response to Folfirinox, lasting for 9 months, but developed progressive disease and died 18 months from diagnosis. In his treatment-naïve primary PanNET, a heterozygotic SPV (p.Ala357fs, VAF 49%) was identified, and interestingly, we detected LOH of the MUTYH wild-type allele (VAF 86%) in his synchronic metastatic PanNET, with the SBS36 mutational signature.
Six patients received immune checkpoint inhibitors. One patient had a PanNET with low TMB and experienced upfront progression with pembrolizumab; another patient with a low TMB had disease controlled for 13 months with pembrolizumab combined with lenvatinib. A patient whose TMB was unknown had upfront NET progression with pembrolizumab. Three patients were treated with immune checkpoint inhibitors when their tumors became hypermutated, and two had tumor response. These two patients experienced complete and ongoing radiological response (lasting for 14 and 24 months so far) with nivolumab and ipilimumab. The patient with a TMB of 97 had a MUTYH SPV with VAF of 5% in ctDNA (#4); tumor tissue sequencing confirmed hypermutation and demonstrated an SBS11 genetic signature (alkylating-induced), but did not evidence MUTYH SPV. The SPV found ctDNA was likely subclonal. She initially had minor tumor response with pembrolizumab for nearly 6 months, when the tumor progressed; she then presented complete remission with nivolumab and ipilimumab for 18 months so far. The patient with a TMB of 65 and complete response with nivolumab and ipilimumab presented a double hit of MUTYH in the tumor (#19). The other patient (#13) with hypermutated PanNET had a MUTYH SPV with a VAF of 24% (germline testing was not performed) and developed a TMB of 728 after 18 months of CAPTEM; the tumor was unresponsive to pembrolizumab.
Discussion
In this international multicenter retrospective series, we report the first clinical characterization of patients with PanNET and pathogenic variants of MUTYH. Their frequency seems rare, most patients were young, male, and had metastatic disease at diagnosis. MUTYH p.Gly396Asp and p.Tyr179Cys were the most common variants, all being GPV. Of six patients who harbored a second hit event in MUTYH in their tumors, five presented aggressive disease. Overall, nearly two-thirds of metastatic patients demonstrated an aggressive course over time, with NEC-like behavior and rapidly progressive disease; 64% of patients with initial G1–2 PanNET with biopsies upon progression developed G3. TMB high was identified in three out of seven cases pretreated with alkylating agents and/or PRRT. Of these, two experienced complete disease remission with nivolumab and ipilimumab.
Scarpa et al. 4 found 11 germline variants in MUTYH, of which 5 were considered GPV. A novel G:C > T:A somatic mutational signature was discovered and named MUTYH signature (SBS36) after validation in PanNET and an MUTYH-associated polyposis-related tumor, all tumor samples with biallelic inactivation in tumor tissues. Among consecutive Brazilian patients with early-onset NEN, nearly 16% presented a GPV in a cancer predisposing gene, with the most commonly affected gene being MUTYH in four cases. 6 In a large pan-solid tumor cohort of over 229,000 solid tumors, MUTYH alterations were found in 2.8% of tumors, 55% of them predicted to be germline. 13 Recently, two studies also found monoallelic GPV in this gene in patients with PanNET: 3 out of 112 cases in one cohort (two cases with p.Tyr165Cys and one with p.Thr474Profs*3 (ENST00000372115.3)), with all three cases annotated as “biallelic” in SignalDB, likely with LOH, 14 and two cases with MUTYH GPV out of 88 cases in another study. 5 In our cohort, MUTYH mutation p.Gly396Asp, a known pathogenic variant, was the most frequently identified, and this variant has also been associated with small bowel NET.15,16 We also found a low frequency (2.9%) of MUTYH pathogenic variants among 402 screened patients.
The molecular profiling of treatment-naïve MUTYH+ PanNET in our study seems similar to sporadic PanNET,3,4 with MEN1, DAXX, and ATRX mutations being the most commonly found. A recent study reported that MUTYH-altered versus -wild-type cancers had significantly higher TMB and more frequent alterations in KRAS G12C, but not in KRAS in general. 13 In our study, most patients had a TMB below 10 before any treatment, but we found a high TMB in three patients after receiving alkylating agents.
Clinically, several characteristics of PanNET patients with MUTYH pathogenic variants seem peculiar. Median age was 48 years old, whereas the most recent epidemiology data report that nearly 90% of patients with PanNET are 45 years or more, with peak incidence among patients of 70–75 years. 1 While male patients usually comprise 50%–55% in overall PanNET cohorts, in our study, nearly two-thirds were male. 1 One-third of cases had an initial G3 tumor, which seems a high proportion compared to PanNET in general, where G3 is observed in about 12% of cases. 17 The median initial KI-67 of 12% also seems higher than usually observed in PanNET, which is in the range of 5%. 18 The majority of G1–2 NET developed into G3 NET upon progression which is highly unusual. Retrospective series of G1–2 PanNET patients with tumor biopsies performed at progression have reported median KI-67 index increase of 14%–45%. 18 The largest series evaluating PanNET grade progression observed that only 12 (4.5%) out of 264 samples of initially G1–2 changed to a G3 PanNET. 19
During the past two decades, the genetic profiles of PanNET have been investigated and researchers have tried to identify if genetic markers influenced prognosis. MEN1, DAXX, and ATRX mutations are the most commonly found in PanNET.3,4 Yet, their correlation with patient prognosis has not been elucidated. Some authors have associated those mutations with improved OS in the metastatic setting.3,5,20 Another retrospective study found the opposite: patients with PanNET carrying MEN1, DAXX, and ATRX mutations had worse clinical outcomes than those with tumors carrying the wild-type alleles of all three genes; the authors suggested that the presence of these mutations was associated with an alpha-cell signature, similarly to the alpha-cells of pancreatic islets, and related to a more aggressive behavior. 21 BRAF and SETD2 alterations have also been associated with a more aggressive behavior in PanNET. 5 Ours is the first study to suggest that either GVP and/or SPV of MUTYH predispose to worse prognosis in metastatic PanNET patients.
The majority (63%) of MUTYH+ PanNET had an aggressive course, with clinical NEC-like behavior (and NEC morphology when tumor samples were examined), either at diagnosis or after progression. While median OS of 4.6 years is similar to that reported in clinical trials 22 and population-based studies 1 of G1–2 metastatic PanNET patients, 7 (58%) out of 12 cases classified as aggressive had initially indolent disease which later became aggressive during the disease course. This finding makes OS an inaccurate endpoint of our cohort. Also, the OS of a patient with G3 PanNET and MUTYH GPV in a large retrospective series of G3 NEN was shorter than the OS of G3 PanNET patients with other GPV. 23
Nearly 40% of tumors tested for LOH evidenced a possible second hit in the PanNET. Interestingly, four of six aggressive cases with GPV and tumor profiling had evidence of a second hit genomic event in MUTYH. Five out of six cases with a double hit event in MUTYH in their tumors presented aggressive disease. These findings suggest a possible specific driver mechanism in MUTYH pathogenic variants. It is possible that heterozygous MUTYH GPV cause some degree of DNA repair defects in PanNET, which are boosted by cytotoxic therapy, predisposing LOH of the wild alleles. A second hit event leads to cumulative genomic instability and tumor progression and could explain why patients with initially indolent PanNET developed aggressive disease after alkylating agents and/or PRRT.
Because of our small sample, it was not possible to properly evaluate response to different therapies. Yet, two complete responses to nivolumab and ipilimumab are unusual in metastatic PanNET. These two patients (#4 and #19) had hypermutated PanNET with mutations in mismatch repair genes, what may explain their excellent outcomes. Pembrolizumab was utilized in two cases with TMB high tumors but it was not effective. One patient had upfront tumor progression (#13); the other, with a MUTYH SPV (VAF 5%) found in ctDNA experienced stable/minor tumor shrinkage for about 6 months and achieved complete remission with nivolumab and ipilimumab (#4). In a recent case report, a Chinese PanNET patient with a MUTYH GPV (and likely with LOH of the wild allele in the tumor) also experienced a partial radiological response to a programmed death receptor-1 monoclonal antibody. 24 How the presence of MUTYH variants contributes to response to immunotherapies in PanNET remains undetermined.
Our study has limitations. The most relevant is the inherent bias associated with the retrospective nature of our study, what prevented an accurate estimate of the frequency of MUTYH GPV and/or SPV in patients with PanNET. Because of our small sample, variable intervals of imaging tests, and lack of central radiology review, it was not possible to estimate response rate and progression free survival of different therapies. Another important limitation was the unavailability of tumor tissues for all patients to investigate LOH of the MUTYH wild alleles. A central pathology revision of tumor samples was not performed but all cases were diagnosed by expert pathologist in NEN who work at NET reference centers.
Future research on the role of MUTYH in the PanNET should be sought. Our group will evaluate the natural history of MUTYH+ tumors, with subsequent tumor biopsies performed upon disease progression. Such studies could help us understand the molecular mechanisms leading to tumor aggressiveness and, in some cases, hypermutation. Another interesting area for research is how MGMT is expressed in PanNET with MUTYH mutations and how this influences responses to alkylating agents.
PanNET are a heterogeneous disease with variable clinical course, even within the tumor grade subgroups. Efforts are needed to identify prognostic molecular biomarkers to help predict disease aggressiveness and to guide more effective treatment for patients. Based on our findings, we recommend that patients with PanNET be evaluated for GPV and SPV, if possible. If a MUTYH GPV is found, genetic counseling of family members is advised and LOH of the wild allele in the tumor could be investigated. For confirmed double hit cases, close monitoring for the developing of aggressive disease is advised (e.g., clinical and radiological evaluation every 3 months). Additionally, molecular profiling of tumor biopsy collected upon progression on therapies could be informative of TMB. If TMB high is found, a combination of nivolumab and ipilimumab could be considered, if there is access to such therapies.
Conclusion
GPV in MUTYH PanNET seem rare and are more common in males and younger patients when compared with the overall PanNET population. Patients with PanNET and MUTYH GPV tend to have more aggressive and high-grade, NEC-like evolution, when metastatic. LOH of the wild allele seems to be associated with tumor aggressiveness.