
Abstract
Antibody–drug conjugates (ADCs) offer a promising therapeutic approach for various cancers, enhancing the therapeutic window while mitigating systemic adverse effects on healthy tissues. ADCs have achieved remarkable clinical success, particularly in treating breast cancer, becoming a standard therapy across all subtypes, including hormone receptor-positive, human epidermal growth factor receptor 2-positive, and triple-negative breast cancer. Although designed to selectively target antigens via monoclonal antibodies, ADCs can exhibit toxicity in normal tissues, often due to off-target effects of their cytotoxic payloads. Understanding and managing these toxicities according to established guidelines are crucial for enhancing ADC clinical efficacy, minimizing adverse events, and ultimately improving patient outcomes. This review comprehensively examines the toxicities of ADCs employed in breast cancer treatment and explores their management strategies. Furthermore, we investigate novel ADCs beyond trastuzumab deruxtecan and sacituzumab govitecan, evaluating their potential efficacy and corresponding safety profiles.
Plain language summary
Antibody-drug conjugates (ADCs) offer a promising therapeutic approach for cancer, especially breast cancer. These therapies utilize antibody targeting to deliver chemotherapy directly to cancer cells. This approach facilitates more precise treatment, minimizing damage to healthy cells and optimizing the balance between efficacy and adverse effects. ADCs are now frequently utilized across all breast cancer subtypes, including hormone receptor-positive, human epidermal growth factor receptor 2-positive, and triple-negative. Despite their targeted design, ADCs can cause side effects because their cytotoxic payloads may also affect healthy cells. These adverse effects vary depending on the specific ADC and may include damage to healthy tissue. Effective management of these toxicities is crucial for maximizing patient benefit from ADCs while mitigating risks. This review describes the management of adverse effects associated with ADCs commonly used in breast cancer treatment.

Introduction
Antibody–drug conjugates (ADCs) represent a promising therapeutic approach to cancer treatment, and their clinical applications are continuously expanding. ADCs comprise a monoclonal antibody (mAb), a cytotoxic payload (a chemotherapeutic agent), and a linker connecting the mAb and payload (Figure 1(a)). Conjugating the payload to mAb enables ADCs to specifically target cancer cells, improving therapeutic efficacy and minimizing off-target toxicity. 1 Several ADCs have demonstrated remarkable success in treating breast cancer, including hormone receptor-positive, human epidermal growth factor 2 (HER2)-positive, and triple-negative breast cancer (TNBC) subtypes, and are currently used in clinical practice.

Structure of ADC and brief mechanisms of ADC toxicity. (a) Schematic diagram of an ADC comprising a monoclonal antibody, a cytotoxic payload, and a linker. (b) Schematic diagram illustrating on-target, off-tumor toxicity. ADCs could bind the target antigens expressed in healthy normal tissues, and, through target-mediated endocytosis, result in target-dependent toxicities. (c) Schematic diagram of off-target, off-tumor toxicity. ADCs could affect healthy non-target tissues, causing target-independent toxicity. ADCs utilizing cleavable linkers frequently release payloads prematurely in plasma, and these released (free) payloads may be internalized by normal cells through passive diffusion or specific transporters. Furthermore, intact ADCs can be internalized into normal cells through receptor-mediated (Fcγ receptors, neonatal Fc receptor, or C-type lectin receptors) or nonspecific endocytosis. 2
Ado-trastuzumab emtansine (T-DM1) became the first ADC approved by the United States (US) Food and Drug Administration (FDA) for patients with HER2-positive metastatic breast cancer (mBC) in 2013 based on the results of EMILIA trial. 3 In addition, T-DM1 was approved for adjuvant treatment for patients with HER2-positive breast cancer who did not achieve a pathologic complete response following neoadjuvant chemotherapy, including taxane, trastuzumab, and/or pertuzumab in 2019. 4 Trastuzumab deruxtecan (T-DXd), another HER2-targeting ADC, represents the second approved ADC for HER2-positive mBC,5–8 and its indication has expanded to include HER2-low mBC based on the DESTINY-Breast04 trial.9,10 Sacituzumab govitecan (SG), which targets trophoblast cell surface antigen 2 (TROP-2), gained approval for heavily treated patients with metastatic TNBC, 11 and hormone receptor-positive/HER2-negative mBC. 12 Beyond these ADCs, numerous novel ADCs are currently undergoing preclinical and clinical trials, and their role in clinical practice is expected to expand further.
Although ADCs have demonstrated remarkable efficacy against breast cancer, their toxicity poses a substantial challenge. A recent meta-analysis of 169 studies employing ADCs indicated that any grade adverse events (AEs) and grade ⩾3 AEs were reported in 91.2% (95% confidence interval (CI) 90.7–91.7) and 46.1% (95% CI 45.2–47.0) of patients, respectively. 13 Similarly, a meta-analysis of 39 studies evaluating HER2-targeted ADCs found the average incidence of any grade AEs was 98.2%, while grade ⩾3 AEs occurred at an average rate of 47.8%. 14 Furthermore, 10.5% of the AEs resulted in treatment discontinuation. 14
Therefore, understanding and managing the toxicities associated with ADCs are essential for their successful clinical implementation. This article comprehensively reviews the toxicities of ADCs employed in breast cancer treatment and discusses their management. Furthermore, we investigate novel ADCs not yet in clinical use, emphasizing their therapeutic potential and safety profiles. All AEs are graded in this manuscript according to the Common Terminology Criteria for Adverse Events, version 5.0. 15
Mechanisms of ADC toxicity
Roughly, 0.1% of an administered ADC dose accumulates at the targeted tumor site, 16 while the majority remains in circulation or affects non-targeted healthy cells, resulting in off-tumor toxicity. This toxicity can be categorized into target-dependent toxicity (also known as on-target toxicity, Figure 1(b)), resulting from low target expression in healthy cells, and target-independent toxicity (off-target toxicity), arising from premature payload release, nonspecific endocytosis, or ADC receptor-mediated endocytosis (Figure 1(c)). Detailed mechanisms of ADC toxicity were extensively reviewed in previous articles.2,17
All three components of ADCs—mAb, payload, and linker—can contribute to their toxicity, though the payload is primarily responsible for the majority of reported adverse effects.18,19 Similar toxicities have been frequently reported with ADCs utilizing the same payloads. For example, grade ⩾3 anemia, neutropenia, and peripheral neuropathy were frequently observed in ADC with monomethyl auristatin E (MMAE) payloads, such as enfortumab vedotin (EV), brentuximab vedotin, polatuzumab vedotin, and pinatuzumab vedotin. 20 Furthermore, thrombocytopenia and hepatic toxicity were commonly noted with ADC with mertansine (DM1), such as T-DM1. 20 Common payload-related toxicities are summarized in Table 1.
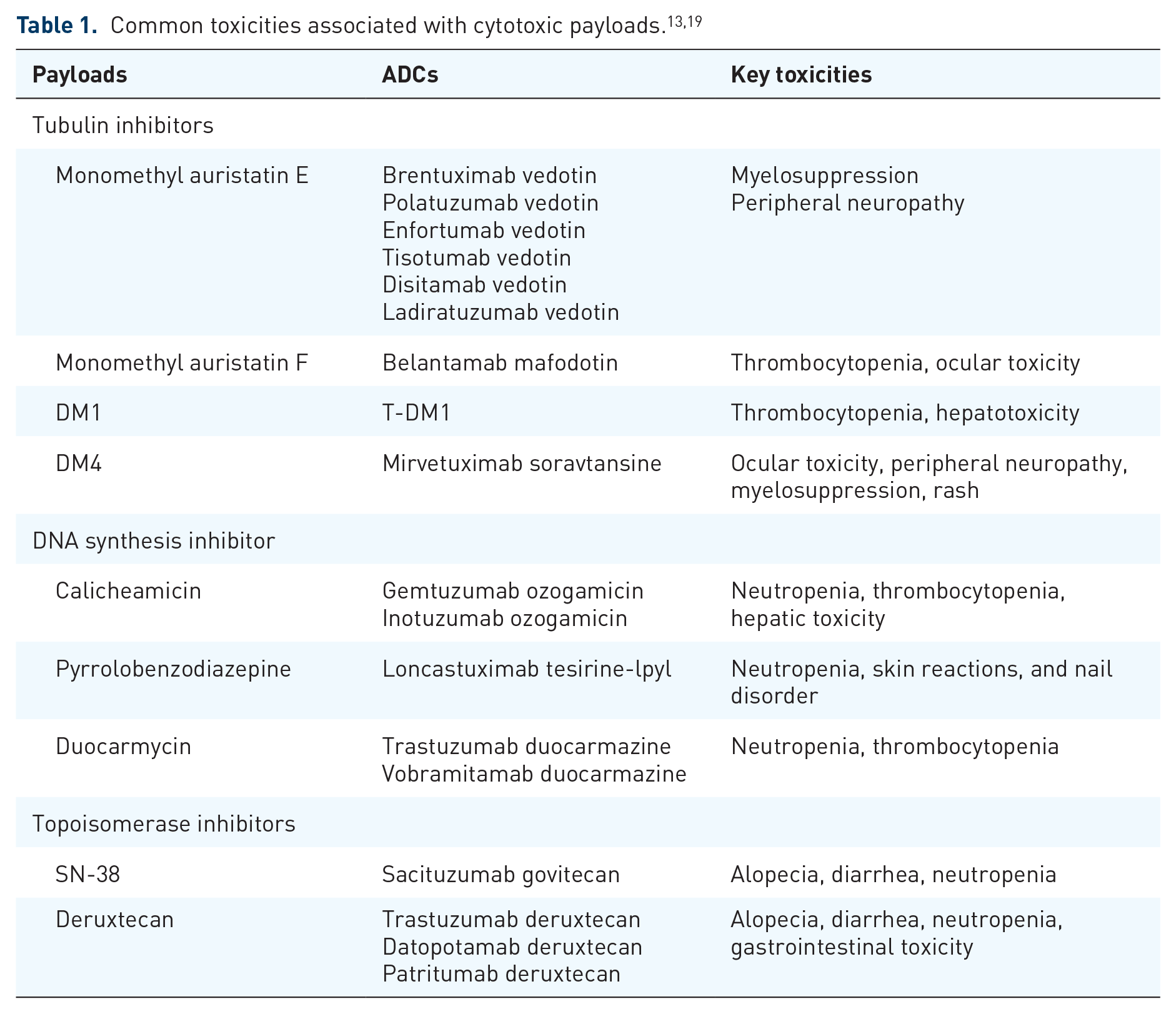
While the linker appears nontoxic, its stability profoundly influences the overall toxicity profile. Linkers are categorized as either cleavable or non-cleavable, depending on the presence of a chemical trigger within their structure. 21 A non-cleavable linker, lacking a chemical trigger, remains stable in systemic circulation until lysosomal release following internalization into target cells. By contrast, cleavable linkers possess a chemical trigger within their structure, enabling cleavage by environmental factors (e.g., pH, lysosomal protease, and glucuronidase) and subsequent payload release in extra-tumoral compartments. 22 This instability enhances ADC efficacy through bystander effects 22 ; however, premature payload release is associated with increased off-target toxicity.2,19,23
Common ADCs used in breast cancer and their associated toxicities and management strategies
Figure 2 illustrates the incidence of common toxicities associated with ADCs used in breast cancer treatment. Table 2 summarizes management strategies for common AEs associated with ADCs, including T-DM1, T-DXd, and SG. Detailed information regarding drug dosing and dose reductions associated with AEs is available in Table 3.

Incidences of common toxicities associated with ADCs in breast cancer treatment. AE incidences are based on the data reported from the EMILIA,3,24 meta-analysis of T-DM1 associated with cardiotoxicity, 25 DESTINY-Breast03, 8 ASCENT, 11 TULIP, 26 ICARUS-BREAST01, 27 and TROPION-Breast01 28 trials. The reported incidence of AEs for the same ADC can vary across different trials.
Management of adverse events associated with T-DM1, T-DXd, and SG.
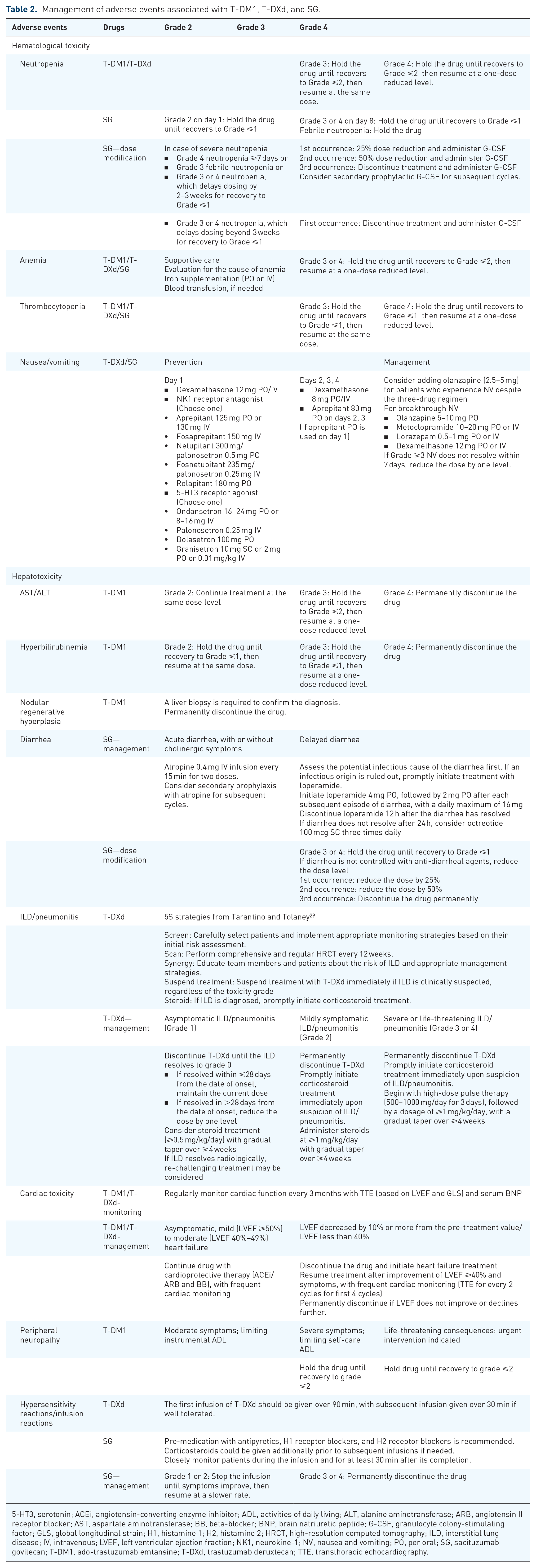
5-HT3, serotonin; ACEi, angiotensin-converting enzyme inhibitor; ADL, activities of daily living; ALT, alanine aminotransferase; ARB, angiotensin II receptor blocker; AST, aspartate aminotransferase; BB, beta-blocker; BNP, brain natriuretic peptide; G-CSF, granulocyte colony-stimulating factor; GLS, global longitudinal strain; H1, histamine 1; H2, histamine 2; HRCT, high-resolution computed tomography; ILD, interstitial lung disease; IV, intravenous; LVEF, left ventricular ejection fraction; NK1, neurokine-1; NV, nausea and vomiting; PO, per oral; SG, sacituzumab govitecan; T-DM1, ado-trastuzumab emtansine; T-DXd, trastuzumab deruxtecan; TTE, transthoracic echocardiography.
Dosing of T-DM1, T-DXd, and SG, and dose levels for dose reductions.
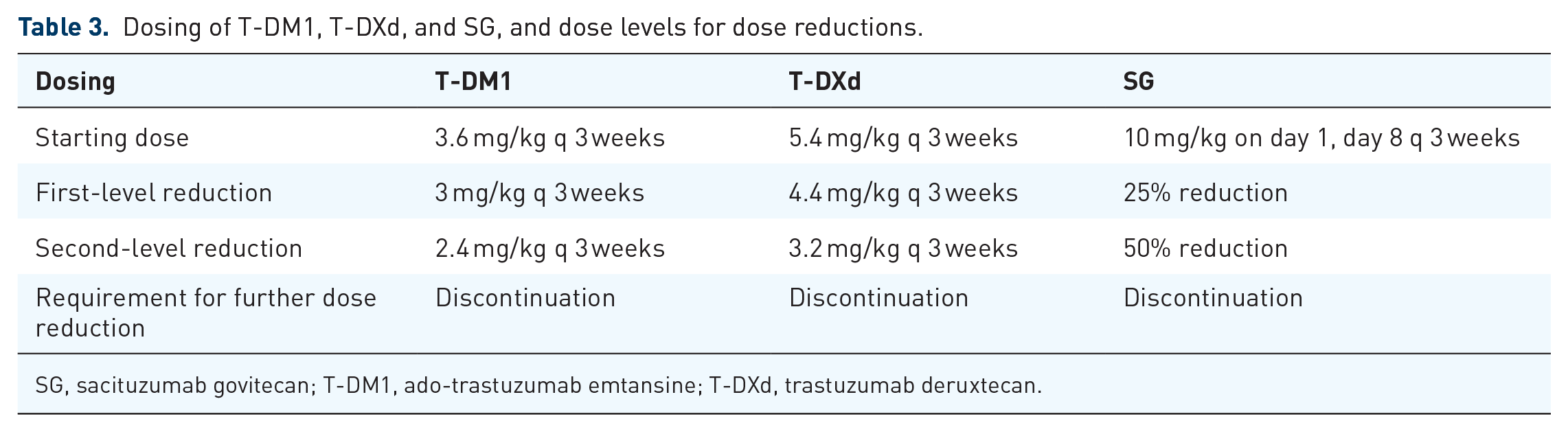
SG, sacituzumab govitecan; T-DM1, ado-trastuzumab emtansine; T-DXd, trastuzumab deruxtecan.
Ado-trastuzumab emtansine
T-DM1 comprises trastuzumab (anti-HER2 mAb), anti-microtubule agents DM1 (a derivative of maytansine as payload), and a non-cleavable linker. 30 T-DM1 is currently used in adjuvant and palliative settings for HER2-positive breast cancer.3,4,24,31 Notable AEs requiring careful monitoring during T-DM1 treatment include thrombocytopenia, hepatotoxicity, cardiotoxicity, peripheral neuropathy, and embryo-fetal toxicity. Because payload (DM1) is metabolized by CYP3A4 and CYP3A5, concomitant administration of strong CYP3A4 inhibitors (e.g., ketoconazole, itraconazole, clarithromycin, and voriconazole) should be avoided due to the potential for increased DM1 exposure and toxicity. 32
Thrombocytopenia
Thrombocytopenia is a dose-limiting toxicity of T-DM1, with grade ⩾3 thrombocytopenia occurring in 6%–15% of patients across phase III studies.3,24,31 Asian patients notably exhibited a higher incidence of grade ⩾3 thrombocytopenia compared to non-Asian patients (44.4% vs 10.6% in pooled analysis 33 ; 36.5% vs 3.7% in KAMILLA study 34 ). However, the incidence of grade ⩾3 bleeding is low, as only 3%–8.1% of patients with grade ⩾3 thrombocytopenia experienced serious hemorrhage, regardless of race.33,34
The mechanisms of T-DM1-induced thrombocytopenia are considered an off-target toxicity affecting megakaryocytes. This condition primarily arises from impaired megakaryocyte differentiation due to T-DM1 internalization via Fcγ receptor-mediated process 35 or macropinocytosis, 36 rather than a direct effect on platelet function. 37 The higher incidence of serious thrombocytopenia in Asian populations may be attributable to specific Fc polymorphisms that are more prevalent in these individuals. 33
Thrombocytopenia was generally managed effectively with dose modifications, without treatment discontinuation.3,34 Monitoring platelet counts before each T-DM1 dose is recommended. For grade ⩾3 thrombocytopenia, withholding T-DM1 until recovery to grade 1 is advised. 38 Empirical platelet transfusion is recommended for patients with platelet counts <10 × 109/L or for those experiencing bleeding with platelet counts <50 × 109/L. 39 The use of thrombopoietin receptor agonists for chemotherapy-induced thrombocytopenia is not approved by the US FDA or the European Medicines Agency. 39
Hepatotoxicity
The mechanisms of T-DM1-induced hepatotoxicity are not well understood, but the payload (DM1) may contribute to on- and off-target toxicity. In mouse models, T-DM1 internalization into hepatocytes occurred via the HER2 receptor, resulting in apoptosis, liver inflammation, and necrosis. 40 By contrast, trastuzumab alone induced only inflammation, not necrosis, in liver tissue, suggesting the payload’s role in hepatotoxicity. 40
Hepatotoxicity is common among patients treated with T-DM1, occurring in 9.2%–28.4% for any grade AE and 0.4%–10.1% for grade ⩾3 AEs.3,4,31,41 A recent meta-analysis demonstrated that T-DM1 treatment (regardless of monotherapy or combination with pertuzumab) increased the risk of any grade and grade ⩾3 transaminitis (risk ratio (RR) 2.73 for grade ⩾3 aspartate aminotransferase (AST) elevation; RR 2.17 for grade ⩾3 alanine transaminase (ALT) elevation). 42 Furthermore, T-DM1 can cause severe liver injury, potentially leading to hepatic failure and even death. 32
Serum AST, ALT, and bilirubin levels should be monitored before initiating T-DM1 treatment and prior to each subsequent dose. In cases of grade 2 AST/ALT elevation, treatment may proceed without dose adjustment. If grade 3 AST/ALT elevation occurs, T-DM1 should be discontinued until recovery to ⩽grade 2, at which point treatment may resume at a reduced dose. Grade 4 elevations in AST/ALT necessitate permanent discontinuation of T-DM1. For grade 2 or 3 hyperbilirubinemia, T-DM1 should be discontinued until recovery to grade ⩽1. Grade 4 hyperbilirubinemia requires permanent discontinuation. 38 In addition, in cases where both AST/ALT exceed three times the upper limit of normal (ULN) and total bilirubin exceeds twice the ULN, T-DM1 should be permanently discontinued (Table 2).
Nodular regenerative hyperplasia (NRH) is a rare liver disease characterized by the diffuse transformation of hepatic parenchyma into small regenerative nodules, which can lead to non-cirrhotic portal hypertension. T-DM1-induced NRH is rare, with few cases documented in the literature.33,43–45 Manifestations of NRH include transaminitis and the insidious onset of portal hypertension (e.g., ascites, variceal bleeding, splenomegaly) without evidence of underlying chronic liver disease. 46 Liver biopsy remains the only definitive diagnostic method for NRH. Upon confirmation of NRH, T-DM1 must be permanently discontinued. NRH management entails addressing the underlying causes and treating complications associated with portal hypertension. 47 In most reported cases, patients recovered after T-DM1 discontinuation or dose reduction.33,44,45,48
Cardiac toxicity
Cardiotoxicity is a well-recognized potential AE of HER2-targeting agents, including trastuzumab, pertuzumab, T-DM1, and T-DXd. Mechanistically, this is considered on-target, off-tumor toxicity associated with HER2-targeting agents. The HER2 receptor, expressed on cardiomyocytes, plays a crucial role in normal fetal heart development and the growth and survival of adult cardiomyocytes.49,50 Preclinical studies demonstrate that ErbB2 deletion in mouse models induces dilated cardiomyopathy and systolic dysfunction. 51 Furthermore, trastuzumab and pertuzumab inhibit a cardioprotective pathway by blocking neuregulin-1-induced ErbB2/ErbB4 heterodimerization. 52
Preclinical studies have shown that T-DM1 induces greater morphological and functional damage to human fetal cardiomyocytes and cardiomyoblasts than trastuzumab, exhibiting effects similar to anthracyclines. 53 In contrast to the preclinical study, which showed a higher toxic effect on cardiomyocytes than trastuzumab, the incidence of cardiotoxicity in patients treated with T-DM1 is relatively low. A recent meta-analysis of advanced HER2-positive breast cancer trials indicated that 3.37% of patients (66 out of 1961) treated with T-DM1 experienced at least one cardiac event, primarily grade 1 or 2 left ventricular ejection fraction (LVEF) reductions (2.04%, n = 40). 25 No patients reported grade ⩾4 toxicity. Moreover, the majority of cardiac toxicity cases (79%) resolved within 1 year. 25 Similarly, the incidence of cardiac events remained low among patients receiving adjuvant T-DM1, with only three patients (0.8%) experiencing grade 3 toxicity. 54 Overall, the incidence of T-DM1 is considered relatively low, typically low grade, and reversible.
No specific guidelines exist for the management of T-DM1-induced cardiotoxicity. Per the 2022 European Society of Cardiology guidelines for cardiotoxicity induced by HER2-targeted agents, regular cardiac function monitoring via transthoracic echocardiography every 3 months, coupled with plasma natriuretic peptide level assessments, is required before and throughout treatment. 55 Cardiac troponins are sensitive markers for detecting cardiotoxicity associated with anthracycline therapy or anthracycline combined with trastuzumab, but not for anti-HER2-targeted agent monotherapy. 56
For patients with asymptomatic mild (LVEF ⩾50%) to moderate (LVEF 40%–49%) heart failure, continued HER2-targeted treatment with cardioprotective therapy (angiotensin-converting enzyme inhibitors/angiotensin II receptor blockers and beta-blockers) is recommended with frequent cardiac monitoring. 55 If the LVEF decreases by ⩾10% from the pre-treatment value or <40%, discontinuation of treatment and reassessment of cardiac function after 3 weeks is recommended by the T-DM1 FDA label. 38 If the LVEF recovers to ⩾40% (ideally 50%) and heart failure symptoms improve, HER2-targeted therapy may be resumed with frequent cardiac monitoring (echocardiography every two cycles for the first four cycles, with the frequency reduced if cardiac function remains stable). 55 If the LVEF does not improve or continues to decline, it is recommended to permanently discontinue the treatment. 38 Heart failure treatment should be initiated in patients with moderate to severe asymptomatic and symptomatic therapy-related cardiac dysfunction. 55
Peripheral neuropathy
Both on-target and off-target toxicities contributed mechanistically to the neurotoxicity induced by T-DM1. The microtubule-inhibiting payload (DM1) causes neurotoxicity through dose-dependent axonal degeneration, suggesting target-independent mechanisms. 57 However, a separate preclinical study indicated potential on-target toxicity because only monkeys (a HER2-binding species) exhibited neurotoxicity after T-DM1 treatment, whereas rats did not exhibit neurotoxicity following T-DM1 or DM1 treatment. 58
The incidence of all grades of peripheral neuropathy in patients treated with T-DM1 was reported to range from 5% to 18.3% in phase III trials, with grade 3 peripheral neuropathy occurring in 0.5%–2.2% of cases.3,4,31 In a meta-analysis, the relative risk of developing peripheral neuropathy was not significantly higher with T-DM1 regimens compared to other therapies in patients with HER2-positive cancers, 59 and it was lower than that associated with treatment with taxane. 60 In addition, peripheral neuropathy was resolved in 74.6% of patients in the KATHERINE trial. 31 Thus, T-DM1-induced peripheral neuropathy is generally considered mild (grade ⩽2) and manageable through dose reduction or temporary treatment discontinuation. For grade ⩾3 peripheral neuropathy, FDA labels recommend discontinuing T-DM1 until recovery to grade 2 or less.
Summary of AEs associated with T-DM1 and recommendations
T-DM1 generally demonstrates a favorable safety profile compared to cytotoxic chemotherapy, with most AEs effectively managed through appropriate mitigation strategies.24,31 Among the AEs, thrombocytopenia and transaminitis are the most common grade 3 events and primary reasons for dose delays or reductions.3,24,31 Hepatotoxicity, cardiac toxicity, and embryo-fetal toxicity are listed by the US FDA as one of the black box warnings of T-DM1 treatment. 38 Regular monitoring of complete blood counts, liver function, and cardiac function is recommended for patients receiving T-DM1, despite the low incidence (<1%) of grade ⩾3 cardiac dysfunction.24,25 Patients should be informed of the risks of severe liver injury, cardiac toxicity, and potential fetal harm. In addition, patients should be advised to seek immediate medical attention if they experience symptoms indicative of acute hepatitis (e.g., nausea, vomiting, jaundice, dark urine, generalized pruritus, or anorexia) or heart failure (e.g., new-onset or worsening shortness of breath, cough, generalized edema, dizziness, or loss of consciousness). 38
Trastuzumab deruxtecan
T-DXd consists of trastuzumab, the topoisomerase I inhibitor deruxtecan (payload), and a tetrapeptide-based cleavable linker. T-DXd is approved for the treatment of HER2-positive and HER2-low mBC.6,7,9 Recently, it also demonstrated clinically meaningful efficacy in patients with HER2-ultralow mBC. 61
In pivotal clinical trials, almost all patients experienced treatment-emergent adverse events (TEAEs), with the incidence of grade ⩾3 TEAEs ranging from 52.6% to 56% (Supplemental Table 1).6,7,9,61 Compared to T-DM1, treatment with T-DXd showed greater rates of drug-related treatment discontinuation (20% vs 7%), dose reduction (25% vs 15%), and dose interruption (42% vs 17%; Supplemental Table 1). 7 Commonly reported AEs (⩾20%) included nausea, vomiting, alopecia, constipation, fatigue, neutropenia, anorexia, anemia, thrombocytopenia, and diarrhea. 5 Interstitial lung disease (ILD) and embryo-fetal toxicity are notable toxicities of T-DXd.
ILD/pneumonitis
ILD/pneumonitis is a particularly noteworthy AE associated with T-DXd. In a pooled analysis of T-DXd clinical trials, the overall incidence of adjudicated drug-related ILD/pneumonitis among patients with breast cancer was 20.6%, with the majority (79%) being grade 1 or 2. Grade 5 toxicity (death) occurred in 2.9% of patients. 62 These results align with a meta-analysis of patients with mBC treated with T-DXd, which reported an 11.7% (95% CI 9.1–15) incidence of ILD, with the majority of cases (80.2%) being mild (grade 1 or 2). 63 Most cases occurred within 12 months of initiation of T-DXd (87%), with a median time to onset of 5.4 months. 62 In the pooled analysis of the DESTINY-Breast 01–04 trials, ILD was recovered or resolved in most patients (62.6%), whereas 18.1% of patients did not experience recovery. 64
The precise mechanism of T-DXd-associated ILD/pneumonitis remains incompletely elucidated. In the preclinical monkey study, lung toxicity presented as diffuse alveolar damage, primarily localized to the alveolar region rather than the pulmonary epithelium following T-DXd treatment. These patterns resemble those observed in human lung toxicity. 65 Since HER2 expression in human and monkey lungs is restricted to the bronchial epithelium, rather than the alveolar epithelium, alveolar cell toxicity may be attributable to target-independent uptake of T-DXd into alveolar macrophages, representing off-target toxicity.65,66 Furthermore, pulmonary toxicity in monkeys exhibited both dose- and frequency-dependent patterns, suggesting a direct cytotoxic effect of deruxtecan rather than immune-mediated mechanisms. 65 Further investigation is warranted to elucidate the mechanisms of T-DXd-related ILD/pneumonitis.
Because ILD/pneumonitis can lead to pulmonary fibrosis and even death, comprehensive screening and prompt management are essential. Building on clinical trial data and real-world experience, Tarantino and Tolaney 29 suggested 5S strategies: “Screen, Scan, Synergy, Suspend treatment, Steroids” for patients receiving T-DXd. Critically, prior to initiating T-DXd, careful patient selection is essential to ensure personalized monitoring strategies based on individual risk profiles. Notable suspected risk factors include age (<65 years), ethnicity (Japan), the presence of lung comorbidities, moderate to severe renal impairment (creatinine clearance <60 ml/min), and low baseline oxygen saturation (<95%).62,64 Moderate renal impairment at baseline correlated with higher rates of ILD/pneumonitis and earlier onset. 64 Lung metastases nor lymphangitic carcinomatosis at baseline, nor prior radiotherapy to the chest or lung, correlated with ILD/pneumonitis in the pooled analysis. 62 Regular clinical assessments are recommended at each visit during treatment, including monitoring oxygen saturation levels.
Second, regular and comprehensive scans are recommended. A baseline high-resolution computed tomography (HRCT) scan of the chest is recommended prior to treatment initiation. During the first year, all patients require the HRCT scan at least every 12 weeks, with those at higher risk (e.g., Asians and those with low oxygen saturation) requiring more frequent imaging. To minimize the risk of ILD and ensure its prompt management, education of both healthcare providers and patients regarding ILD risk factors and associated symptoms (e.g., dry cough, dyspnea, and fever; synergy) is essential. Patients should be advised to promptly report any new or worsening symptoms to their physician.
Third, if ILD/pneumonitis is clinically suspected, T-DXd should be discontinued immediately, irrespective of toxicity grade. Evaluation should encompass HRCT, consultation with a pulmonologist, pulse oximetry, and diagnostic tests to differentiate infection or metastasis. If ILD/pneumonitis is diagnosed, corticosteroids are the cornerstone of treatment, with dosages adjusted according to the toxicity grade: grade 1, consider administering 0.5 mg/kg/day; grade 2, immediate initiation of 1 mg/kg/day; and grades 3 or 4, 500–1000 mg/day for 3 days, followed by ⩾1 mg/kg/day. 67 Steroids should be maintained for at least 14 days or until clinical recovery, followed by a gradual taper over 4 weeks. Prophylaxis for Pneumocystis jirovecii pneumonia should be considered for patients receiving corticosteroids at a dose of >30 mg/day for >4 weeks. 68
Re-challenge with T-DXd following ILD/pneumonitis should be undertaken with caution. The current guidelines restrict re-challenge to patients with completely resolved grade 1 ILD. 69 If ILD/pneumonitis resolves within 28 days, the dose level may be maintained. Upon recovery after 28 days, the prescribed dose should be lowered. 70
In a pooled analysis of 47 patients with grade 1 ILD/pneumonitis re-challenged with T-DXd, only three patients (6%) experienced recurrent ILD/pneumonitis. 62 Recently, Rugo et al. reported similar findings in a re-challenge study of T-DXd in patients with grade 1 ILD/pneumonitis (n = 45). Consequently, 33.3% of patients (15 out of 45) experienced recurrent ILD and 20% (9 out of 45) discontinued treatment due to recurrent ILD. Notably, all recurrent ILD events were mild and generally responsive to current management guidelines. 71 While some case studies have reported successful re-challenge after grade 2 or 3 ILD/pneumonitis,72,73 permanent discontinuation of T-DXd is recommended for patients with grade ⩾2 toxicity in current guidelines. 69 Further prospective and real-world studies are warranted to investigate the potential for T-DXd re-challenge in patients who experienced grade ⩾2 ILD/pneumonitis.
Nausea and vomiting
Effective prevention and management of nausea and vomiting (NV) are crucial for maintaining a patient’s quality of life and treatment adherence. T-DXd is classified as a high emetic risk drug by the National Comprehensive Cancer Network (NCCN) guidelines. 74 In a pooled analysis of seven clinical trials with patients treated with T-DXd 5.4 mg/kg, 74.6% and 41.6% of patients experienced any grade of NV, respectively. Patients experienced grade ⩾3 NV at rates of 5.8% and 2.6%, respectively. 75
A three-drug regimen (combination of neurokine-1 receptor antagonist, serotonin receptor antagonist, and dexamethasone) is recommended to prevent acute (before 24 h of infusion) and delayed-onset (after 24 h of infusion) NV in patients treated with T-DXd. For patients experiencing NV despite the three-drug regimen, adding olanzapine (2.5 or 5 mg) on days 2–4 is recommended. 74 If grade ⩾3 nausea persists beyond 7 days, dose reduction by one dose level is recommended. 76 Most cases of nausea can be effectively managed with prophylaxis protocol (Table 2).
Hematological toxicity—Neutropenia, anemia, and thrombocytopenia
Hematological toxicity frequently occurs during T-DXd therapy. Any grade neutropenia and anemia occurred in 31%–34.8% and 29.9%–33.2% of patients, respectively.5,6,9 Grade ⩾3 neutropenia and anemia were observed at rates of 6%–19.6% and 8%–9%, respectively.5,6,9 Febrile neutropenia, in contrast, was infrequent, reported in 1.6% and 0.3% in DESTINY-Breast01 5 and DESTINY-Breast04 9 trials, respectively. In a subgroup analysis of the DESTINY-Breast03 trial, neutropenia and anemia were more prevalent among the Asian population than the overall population. 77 Thrombocytopenia was reported less frequently with T-DXd compared to T-DM1 in the DESTINY-Breast03 trial (25% vs 44% for any grade; 8% vs 20% for grade ⩾3). 7
Baseline complete blood count monitoring is recommended before initiating T-DXd and prior to each subsequent dose. Neutropenia can be mitigated through intermittent treatment interruptions and dose reductions. In cases of grade 3 neutropenia, chemotherapy is withheld until recovery to grade ⩽2, at which point it can be resumed at the original dose. For grade 4 neutropenia, defer chemotherapy until recovery to grade ⩽2 and then resume the drug at a dose reduced by one level. Routine primary prophylaxis with granulocyte colony-stimulating factor (G-CSF) is not recommended for patients receiving T-DXd due to the low incidence of febrile neutropenia. However, primary prophylaxis with G-CSF may be considered for patients with high-risk factors (e.g., age ⩾65 years, advanced disease stage, and prior febrile neutropenia with chemotherapy).78,79 Secondary G-CSF prophylaxis may be considered for patients experiencing grade 3 neutropenia who require a rapid antitumor response, thereby preventing dose delays and maximizing the benefit of T-DXd. Administration options include short-acting G-CSF for 2–3 days or pegylated G-CSF on day 2.78,80
Others—Cardiac toxicity and embryo-fetal toxicity
Treatment with anti-HER2-targeted agents is linked to the development of cardiomyopathy. In the DESTINY-Breast01 and DESTINY-Breast03 studies, the incidence of decreased LVEF was reported as 1.6% and 2.3%, respectively.5,7,8 Furthermore, the majority of events resolved spontaneously. Conversely, in the DESTINY-Breast04 study, 11.4% of patients (n = 44) exhibited decreased LVEF, 4.6% (n = 17) presented with left ventricular dysfunction, and 2 patients developed cardiac failure. A meta-analysis revealed an overall incidence of 1.95% (95% CI 0.65–3.73) for LVEF reduction and 7.77% (95% CI 2.74–20.11) for QT interval prolongation. 63 Therefore, frequent cardiac monitoring is necessary for patients receiving T-DXd. The management of cardiotoxicity associated with T-DXd is comparable to that of T-DM1, as detailed in Table 2.
Administration of T-DXd to pregnant women may cause fetal harm. Trastuzumab is known to cause fetal pulmonary hypoplasia, skeletal abnormalities, and neonatal death. 81 Furthermore, deruxtecan (the payload) can cause embryo-fetal toxicity due to its mechanisms of action, which involve DNA replication and chromatin remodeling. 82 Women of reproductive potential should be advised to use effective contraception during T-DXd treatment and for 7 months following the final dose, while their partners should use contraception during treatment and for at least 4 months after the final dose. 83
Summary of AEs associated with T-DXd and recommendations
The most common TEAEs associated with T-DXd were gastrointestinal symptoms (NV) and hematologic toxicities, which are generally manageable through appropriate mitigation strategies. Prophylactic antiemetics are essential for preventing NV induced by T-DXd, as recommended by established guidelines. Hematological toxicities are generally well-controlled through dose adjustments, such as delays or reductions. Prophylactic G-CSF administration may be considered for patients at high risk. ILD/pneumonitis is an AE of special concern with T-DXd. Although the incidence of drug-related ILD/pneumonitis is relatively low, it represents the leading cause of drug discontinuation and can be fatal in some cases. 7 Therefore, vigilant monitoring and prompt intervention are essential for all patients receiving T-DXd. Patients should be thoroughly informed about the risks of severe or fatal ILD, neutropenia, cardiac dysfunction, and potential fetal harm. Patients should be advised to promptly contact their healthcare provider if they experience symptoms suggestive of ILD/pneumonitis (e.g., cough, shortness of breath, fever, or other new or worsening respiratory symptoms), febrile neutropenia (e.g., fever, any signs of infection), or cardiac dysfunction (e.g., shortness of breath, or generalized edema).
Given the demonstrated efficacy and widespread use of T-DXd, its clinical applications are expected to expand substantially with ongoing clinical trials exploring its potential in broader patient populations (e.g., DESTINY-BREAST12 (NCT04739761), DESTINY-BREAST15 (NCT05950945)). However, the mechanisms underlying T-DXd-related AEs, especially ILD, remain insufficiently understood. Further research elucidating the mechanisms of these AEs is warranted to maximize the therapeutic benefit of T-DXd and enhance patient outcomes.
Sacituzumab govitecan
SG comprises a TROP-2 antibody, an SN-38 (payload), and a hydrolyzable linker.84,85 SN-38 is an active metabolite of the topoisomerase 1 inhibitor irinotecan. 86 SG has received approval for metastatic TNBC and hormone receptor-positive/HER2-negative mBC. In pivotal trials, patients receiving SG experienced a higher incidence of grade ⩾3 TEAEs, serious TEAEs, and drug-related dose interruptions than those receiving chemotherapy. However, the rates of dose reduction or discontinuation were similar, less than 10% in both the SG and chemotherapy groups (Supplemental Table 2).11,12,87 The most reported AEs (⩾20%) of SG are neutropenia, anemia, diarrhea, nausea, vomiting, fatigue, alopecia, anorexia, and nervous system disorder.11,12 Notable AEs associated with SG of grade ⩾3 were neutropenia (51%), diarrhea (10%), anemia (8%), and febrile neutropenia (6%). 11 The incidences of AEs did not differ among patients across various subgroups, including older age (⩾65 years), black race, and individuals with brain metastases. 88 In addition, no differences were observed in clinical outcomes between patients who experienced grade ⩾3 neutropenia or grade ⩾2 diarrhea and those who did not. This suggests that monitoring and managing AEs are crucial for sustained clinical benefit and prolonged treatment duration. 89
The enzyme uridine diphosphate glucuronosyltransferase 1A1 (UGT1A1) plays a critical role in inactivating SN-38 through glucuronidation. 90 Among the patients with UGT1A1 polymorphism (*28 allele), the incidence of neutropenia and diarrhea is high with irinotecan therapy due to reduced activity of UGT1A1. 90 Thus, UGT1A1 genotyping and dose reduction of irinotecan is recommended for those with UGT1A1 polymorphisms when treating metastatic colon cancer with irinotecan. 91 Because SN-38 is the active metabolite of irinotecan, patients treated with SG who have the UGT1A1 homozygous genotype (*28/*28) demonstrate a higher incidence of grade ⩾3 neutropenia, diarrhea, and febrile neutropenia compared to those with the heterozygous genotype (*1/*28) and the wild-type genotype (*1/*1) (58% vs 47% vs 53% for grade ⩾3 neutropenia; 15% vs 9% vs 10% for grade ⩾3 diarrhea; 18% vs 5% vs 3% for febrile neutropenia), respectively. 92 Similarly, Wong et al. 93 reported that patients treated with SG who have UGT1A1 polymorphism (*28/*28) have an increased risk of treatment discontinuation due to toxicity (hazard ratio (HR) 5.52 (95% CI 1.15–26.49)) compared to those with the wild-type genotype. 93 Currently, no specific guidelines exist for managing UGT1A1 polymorphisms in patients receiving SG, especially regarding prescreening or dose adjustments before treatment initiation. However, patients with the UGT1A1 polymorphism require close monitoring for severe neutropenia and diarrhea.
Neutropenia and febrile neutropenia
Neutropenia is the most common AE in phase III trials of SG, primarily attributed to its payload (SN-38). In the ASCENT trial, neutropenia occurred in 63% of patients, with 51% experiencing grade ⩾3. 11 Similarly, in the TROPiCS-02 trial, 70% of patients developed neutropenia, with 51% experiencing grade ⩾3. 12 The incidence of febrile neutropenia was 6% and 5% in the ASCENT and TROPiCS-02 trials, respectively.11,12
If grade 2 neutropenia occurs on day 1 or grade 3 neutropenia occurs on day 8, deferral of SG treatment is recommended until recovery to grade ⩽1. Upon the first occurrence of severe neutropenia (grade 4 neutropenia lasting ⩾7 days, grade 3 or 4 neutropenia resulting in a 2- to 3-week dosing delay or febrile neutropenia), treatment is delayed until recovery to grade 1, at which point the dose is reduced by 25%. Upon a second occurrence, the dose is reduced by 50%, and upon a third, SG treatment should be discontinued.94,95 Furthermore, secondary prophylaxis with G-CSF may be considered for patients experiencing severe neutropenia (Table 2). 95
In the phase II PRIMED trial, patients treated with SG for the first two cycles received prophylactic short-acting G-CSF and loperamide to assess whether this primary prophylaxis could improve SG tolerability. 96 After two cycles, 16% of patients (n = 8) experienced grade ⩾3 neutropenia, demonstrating the efficacy of short-acting G-CSF in preventing severe neutropenia (p = 0.0002). While current guidelines do not recommend primary G-CSF prophylaxis, the findings from the PRIMED trial indicate its potential efficacy and merit further investigation. Extensive studies are needed to validate these findings and establish optimal primary prophylaxis strategies for severe neutropenia, including G-CSF administration timing (day 1 or 8) and formulation (short-acting or pegylated G-CSF).
Diarrhea
Diarrhea is another common AE associated with SG and is included in the FDA’s boxed warnings. 94 Severe diarrhea can cause electrolyte imbalances, dehydration, and even acute kidney injury, necessitating prompt treatment. The incidence of any grade and grade ⩾3 diarrhea was reported as 59% and 10%, respectively, in the ASCENT trial and 57% and 9%, in the TROPiCS-02 trial.11,12 In the ASCENT trial, the median time to onset of the first episode of grade ⩾3 diarrhea was 19 days, and the median duration was 5 days. 92
SN-38 is the primary cause of SG-related diarrhea, principally due to its induction of oxidative stress and subsequent damage to the intestinal mucosa.97–99 Preclinical models have demonstrated a correlation between intestinal SN-38 concentrations and diarrhea severity. 100 Early dissociation of SN-38 from SG is thought to be a key mechanism of SG-related diarrhea, representing off-target toxicity.101,102
Diarrhea can manifest as either an acute or a delayed condition. For acute diarrhea, with or without early cholinergic symptoms (e.g., abdominal cramping, sweating, or excessive salivation) during or shortly after SG infusion, administer atropine 0.4 mg intravenously every 15 min for two doses. Furthermore, secondary prophylaxis with atropine is recommended for subsequent treatment cycles.94,95,101 For grade 1 or 2 delayed diarrhea, loperamide treatment is recommended after excluding infectious diarrhea. Patients can initially receive 4 mg of loperamide orally, followed by 2 mg after each subsequent diarrheal episode. The maximum daily dose is 16 mg, and the risk of overdose is low due to minimal systemic absorption. After the diarrhea is resolved, loperamide can be discontinued 12 h after the last episode. If diarrhea persists for more than 24 h, consider subcutaneous octreotide at 100–150 mcg three times daily. 103 For grade ⩾3 diarrhea, hospitalization, and intravenous fluid replacement are recommended.94,95
Prophylactic loperamide is not routinely recommended. 95 However, the PRIMED study showed the efficacy of prophylactic loperamide (2 mg per oral twice daily or 4 mg per oral once daily on days 2, 3, 4, 9, 10, and 11) in preventing SG-induced diarrhea, with grade ⩾2 diarrhea occurring in only 8% of patients treated with SG for two cycles. While these results lack statistical significance (p = 0.0084), they suggest a potential benefit to prophylactic loperamide use and underscore the need for a larger, prospective study. 96
Hypersensitivity- and infusion-related reactions
Hypersensitivity reactions (e.g., wheezing, angioedema, swelling, pneumonitis, skin reactions, hypotension, or cardiac arrest) are common during SG treatment. In the ASCENT trial, any grade and grade ⩾3 AEs occurred in 34.1% and 1.7% of patients, respectively. 11 Prophylactically, antipyretics and histamine H1 and H2 receptor blockers are recommended. If necessary, supplemental corticosteroids may be administered. Patients should be closely monitored for at least 30 min during and after the initiation of SG.94,95 If grade 1 or 2 hypersensitivity reactions occur, the infusion should be paused until symptom improvement. After the patient stabilizes, the infusion rate may be reduced and resumed. For grade ⩾3 hypersensitivity reactions, SG should be permanently discontinued.11,94
Nausea and vomiting
According to NCCN guidelines, SG is classified as a highly emetogenic drug. 74 In phase III clinical trials, the reported incidence of any grade nausea was 55%–57%, with grade ⩾3 nausea occurring in 1%–2% of patients. The incidence of any grade vomiting ranged from 19% to 29%, with grade ⩾3 vomiting occurring in 1% of patients. The management strategies are similar to those for T-DXd.
Embryo-fetal toxicity
SN-38, a genotoxic compound, can cause teratogenicity and/or embryo-fetal lethality in pregnant women.104,105 Therefore, female patients of reproductive potential should be advised to use effective contraception during treatment and for 6 months after the final dose. Male patients with female partners of childbearing potential should use effective contraception during treatment and for 3 months after the final dose. 94
Summary of AEs associated with SG and recommendations
The most common AEs associated with SG include hematologic toxicities (neutropenia and anemia) and gastrointestinal symptoms (diarrhea and NV). The US FDA has issued a black box warning for neutropenia and diarrhea due to the potential risk of severe or life-threatening outcomes. The incidence of these AEs is higher in patients homozygous for the UGT1A1*28 polymorphism. Management of these conditions may necessitate SG dose adjustments based on AE severity and may include prophylactic medications, such as G-CSF, loperamide, or atropine. Patients should contact their healthcare provider if they experience symptoms suggestive of febrile neutropenia (e.g., fever, chills, or other signs of infection) or severe diarrhea (e.g., black or bloody stools, dehydration symptoms such as light-headedness, dizziness, or faintness, or uncontrolled diarrhea lasting more than 24 h). Moreover, patients should be counseled on the risks of infusion-related reactions and the potential teratogenic effects of SG, underscoring the importance of appropriate precautions to mitigate fetal harm.
New emerging ADC in the era of T-DXd and SG
Trastuzumab duocarmazine
Trastuzumab duocarmazine (T-duo, SYD985) comprises trastuzumab and duocarmycin prodrugs (seco-DUBA) payloads linked by a cleavable peptide linker.106,107 Duocarmycin binds to DNA minor grooves and alkylates adenine, inducing DNA strand breaks. 108 This mechanism enables it to target both actively proliferating and quiescent cancer cells, ultimately leading to cell death. T-duo has a drug-to-antibody ratio of 2.8. Similar to T-DXd, T-duo exhibits bystander killing effects due to a cleavable linker. 109 In a phase I study, 33% of patients (16 out of 48) with HER2-positive mBC and 34% of patients (15 out of 43; with 9 hormone receptor-positive and 6 hormone receptor-negative) with HER2-low mBC demonstrated a partial response to T-duo. 110 In a phase III TULIP trial, T-duo showed a significant improvement of progression-free survival (PFS) in patients with pre-treated HER2-positive mBC compared to physician’s choice treatment (7.0 vs 4.9 months, HR 0.63 (95% CI 0.49–0.84, p = 0.002)); however, the difference in overall survival (OS) was not statistically significant (p = 0.236). 26 The most common AEs (⩾20%) were conjunctivitis (38.2%), keratitis (38.2%), fatigue (33.7%), dry eye (30.2%), nausea (25.7%), alopecia (21.5%), diarrhea (20.8%), and asthenia (20.1%). Significant AEs included ocular toxicity and ILD/pneumonitis. Cardiotoxicity is also a concern with T-duo due to trastuzumab. The incidence of AEs related to T-duo is summarized in Table 4 and Supplemental Table 3.
Emerging ADC structures and common AEs in breast cancer treatment.

ADC, antibody–drug conjugates; AE, adverse events; ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; DAR, drug-to-antibody ratio; HER2, human epidermal growth factor receptor 2; ILD, interstitial lung disease; MMAE, monomethyl auristatin E; TROP-2, trophoblast cell surface antigen 2.
Ocular toxicity
In a phase I dose-escalation study, 71% of patients (n = 104) experienced one or more ocular AEs, while 7% (n = 10) experienced grade 3 events. Patients typically experienced grade 3 toxicity after a median of 7.6 months. 110 In the phase III TULIP study, any grade of ocular toxicity (conjunctivitis, keratitis, dry eye, and increased lacrimation) occurred in 78.1% of patients treated with T-duo, compared to 29.9% in the control arm. Grade ⩾3 ocular toxicity presented in 21.2% of patients treated with T-duo. Furthermore, 22.9% of patients underwent dose modification, while 21.5% discontinued treatment due to ocular toxicity. 26
The mechanism underlying ocular toxicity remains poorly understood.110,116 One potential mechanism involves HER2 expression in the human corneal limbal and conjunctival epithelium, 117 possibly leading to on-target, off-tumor toxicity. However, the incidence of ocular toxicity varies among HER2-targeting agents, with T-duo exhibiting the highest rate of ocular AEs. 118 This suggests that T-duo’s ocular toxicity cannot be fully attributed to the on-target effects of trastuzumab alone. A meta-analysis revealed a higher incidence of ocular toxicity with ADCs utilizing DM1, DM4, and monomethyl auristatin F (MMAF) payloads, suggesting the involvement of payload in this AE. 20 Further investigation is needed to elucidate the precise mechanisms underlying T-duo-related ocular toxicity and subsequently refine prevention and treatment strategies.
In a phase I study, most ocular toxicities were resolved with medical interventions, such as eye drops or ointment. 110 Prophylactic lubricating eye drops, regular ophthalmologic examinations, and consultations with ophthalmologists comprised the mitigation strategies. 110 In the case of grade ⩾3 keratitis, treatment should be discontinued.
ILD/pneumonitis
In a phase I dose-escalation study, 8% of patients (3 out of 39) developed grade 1 or 2 pneumonitis, with one fatality attributed to this condition. All events occurred at ⩾1.5 mg/kg doses. 110 In the phase III TULIP study, the incidence of all-grade ILD/pneumonitis was 7.6%, with a grade ⩾3 incidence of 2.4%. Furthermore, 5.2% of patients discontinued treatment due to ILD/pneumonitis, while 2.1% required dose adjustments. 26 Similar to T-DXd, T-Duo is based on trastuzumab, which may be a contributing factor to the on-target toxicity of ILD/pneumonitis. However, the precise mechanisms underlying T-duo-related ILD/pneumonitis remain elusive. Per protocol, mitigation strategies, HRCT scans, and diagnostic work-ups are recommended for new or worsening respiratory symptoms. 26 In the case of grade ⩾2 pneumonitis, treatment should be discontinued. 26
Datopotamab deruxtecan
Datopotamab deruxtecan (Dato-DXd) is an ADC comprising a TROP-2-directed mAb, deruxtecan (a topoisomerase I inhibitor), and a tumor-selective tetrapeptide-based cleavable linker. 119 In the phase I TROPION-PanTumor01 study, the confirmed objective response rates (ORRs) were 26.8% and 31.8% in heavily treated patients with hormone receptor-positive/HER2-negative breast cancer and TNBC, respectively. 120 Furthermore, in the phase III TROPION-Breast01 study, Dato-DXd improved PFS compared to the investigator’s choice of chemotherapy (eribulin, vinorelbine, capecitabine, or gemcitabine) in previously treated patients (one or two lines) with hormone receptor-positive/HER2-negative breast cancer (6.9 vs 4.9 months, HR 0.63 (95% CI 0.52–0.76), p < 0.001). 28 Furthermore, the incidence of grade ⩾3 treatment-related AEs (TRAEs) of Dato-DXd was less than half of those observed in the control group (20.8% vs 44.7%), indicating better tolerability of Dato-DXd. 28 Multiple clinical trials are evaluating Dato-DXd in patients with breast cancer, notably the TROPION-Breast02 study (Dato-DXd vs investigator’s choice chemotherapy as first-line treatment for patients with TNBC, NCT05374512), the TUXEDO-2 trial (in patients with brain metastases, NCT05866432), and the TROPION-Breast05 study (Dato-DXd with or without durvalumab vs investigator’s choice chemotherapy plus pembrolizumab for patients with TNBC, NCT06103864).
The most frequently reported TRAEs (⩾20%) were nausea (51%), stomatitis (50%), alopecia (36.4%), fatigue (23.6%), dry eye (21.7%), and vomiting (19.7%; Table 4). 28 Compared to SG, which targets TROP-2 and utilizes a similar payload (topoisomerase I inhibitor) as Dato-DXd, patients treated with Dato-DXd experienced a low incidence of neutropenia and diarrhea. This difference may be attributable to the stability of the Dato-DXd linker in systemic circulation, ensuring selective payload release through proteolytic processing by enriched lysosomal enzymes within tumor cells and consequently minimizing off-target toxicity. 119 Notable AEs associated with Dato-DXd included stomatitis, ocular toxicity, and ILD/pneumonitis.
Oral mucositis/stomatitis
Oral mucositis/stomatitis is a common toxicity of Dato-DXd. High TROP-2 expression in mucosal tissues is thought to contribute to this on-target toxicity. 121 In the TROPION-PanTumor01 study, the incidence of any grade stomatitis was reported as 79.5%–90.2%, with grade ⩾3 stomatitis occurring in 9.8%–11.4% of patients. 120 One patient (2.4%) discontinued treatment due to grade 3 stomatitis; 14 patients required dose delays, and 4 patients required dose reductions. In the TROPION-Breast01 study, any grade and grade 3 oral mucositis/stomatitis occurred in 55.6% and 6.9% of patients, respectively. 28 No patients experienced grade ⩾4 toxicity, and only one patient (0.3%) discontinued treatment. 28 Overall, most toxicities were mild and effectively managed with appropriate strategies. The higher incidence of oral mucositis/stomatitis observed in the TROPION-PanTumor01 study, compared with the TROPION-Breast01 study, may be attributable to the absence of a prophylactic strategy. 120
Daily prophylactic use of a steroid-containing mouthwash is recommended as a mitigation strategy. Should a steroid-containing mouthwash be unavailable, daily rinsing with a non-alcoholic or bicarbonate-containing mouthwash offers an alternative. Prophylactic cryotherapy, such as holding ice chips or ice water in the mouth during infusion, is also recommended. 28 For grade 2 or 3 toxicity, dose delays are recommended until recovery to grade ⩽1 or baseline. 122 For grade 4 toxicity, discontinuation of Dato-DXd is recommended.
Ocular toxicity
In the TROPION-PanTumor01 study, ocular toxicity incidence ranged from 36.4% to 41.5% for any grade, with grade 3 toxicity affecting 2.4% of patients. In the hormone receptor-positive/HER2-negative cohort, 4.9% of patients (n = 2) discontinued Dato-DXd due to ocular toxicity. 120 The TROPION-Breast01 study reported a 40% overall incidence of ocular toxicity, primarily grade 1 (31.9%), with grade 3 events occurring in only 0.8% of patients. Dose reductions or interruptions occurred in 3.3% of patients, while dose discontinuation occurred in 0.3%. The most frequently reported AEs were dry eye (21.7%), keratitis (14.4%), and increased lacrimation (6.4%). 28
The mechanisms of Dato-DXd-related ocular toxicity remain poorly defined. 122 One possible explanation is that TROP-2 expression in the cornea may contribute to the on-target toxicity of Dato-DXd.121,122 However, ocular toxicity is uncommon with SG (which has TROP-2-directed mAb). 94 These findings suggest additional mechanisms underlying Dato-DXd-related ocular toxicity beyond those associated with the mAb or its payload. The relatively stable linker of Dato-DXd may contribute to its ocular toxicity, as ADCs with more stable linkers have demonstrated a higher incidence of ocular toxicity compared with those possessing less stable linkers.111,123,124
Management of ocular toxicity includes using artificial tears and avoiding contact lenses. Furthermore, regular ophthalmologic examinations are advised every three cycles. 28 Dose modification is required for grade ⩾2 toxicity. 122
ILD/pneumonitis
ILD/pneumonitis is a recognized toxicity associated with DXd-containing ADCs. In the TROPION-PanTumor01 study, only one patient (2.4%) in the hormone receptor-positive/HER2-negative cohort developed grade 3 ILD/pneumonitis, leading to treatment discontinuation. No patients in the TNBC cohort experienced ILD/pneumonitis. 120 In the TROPION-Breast01 study, the incidence of all-grade ILD/pneumonitis was 3.3%, with grade ⩾3 toxicity reported at 0.8% (grade 3 for two patients and grade 5 for one patient). Notably, the cause of death for the patient who experienced grade 5 pneumonitis was disease progression, and the investigator assessed the pneumonitis severity as grade 3. The median time to onset of ILD/pneumonitis is 84.5 days, while the median time to resolution is 28 days. Dose reduction, interruption, and discontinuation occurred in 0.3%, 0.8%, and 1.4% of patients, respectively. 125 The incidence of ILD/pneumonitis is relatively low in patients with breast cancer treated with Dato-DXd compared to patients with non-small-cell lung cancer. 126 Collectively, most cases of adjudicated ILD with Dato-DXd appear to be low grade.
The mechanism underlying this toxicity remains poorly elucidated. Pulmonary alveolar toxicity from the deruxtecan payload may cause ILD/pneumonitis, similar to T-DXd. 65 ILD/pneumonitis treatment strategies for Dato-DXd and T-DXd are similar.
Patritumab deruxtecan
HER3, a transmembrane receptor in the HER family, is overexpressed in various cancers, including melanoma, ovarian, colorectal, gastric, and breast cancers, and this overexpression correlates with poor survival. 127 In breast cancer, HER3 is overexpressed in approximately 50%–70% of all subtypes.128,129 HER3 contributes significantly to resistance against PIK3/AKT/mTOR inhibitors,130–132 HER2-targeted therapies, 133 and endocrine therapy,134,135 suggesting that HER3 is a promising therapeutic target in breast cancer.
Patritumab deruxtecan (HER3-DXd), an ADC comprising a HER3-directed mAb, deruxtecan as payload, and a tetrapeptide-based cleavable linker.136,137 In the phase I/II clinical trial, HER3-DXd demonstrated clinically meaningful efficacy among patients with heavily treated advanced breast cancer across various clinical subtypes, irrespective of HER3 expression. 138 The confirmed ORR was 22.6%–42.9%, the median PFS was 5.5–11.0 months, and the median OS was 14.6–19.5 months across these subtypes. 138 Furthermore, the confirmed ORR in patients with brain metastasis was 28.6%. 138
In the SOLTI-1805 TOT-HER3 study, 77 patients with hormone receptor-positive/HER2-negative breast cancer from part A received HER3-DXd (6.4 mg/kg) as neoadjuvant therapy and demonstrated a meaningful ORR of 45.2%, which included 14 complete responses (22.6%). 139 In part B (treated with 5.6 mg/kg), the clinical ORR was 32% (30% in hormone receptor-positive/HER2-negative cohort and 35% in TNBC) for patients treated with HER3-DXd. 140 These findings suggest that HER3-DXd has the potential to broaden treatment options for breast cancer, offering possible benefits in both neoadjuvant and palliative care settings. Several clinical trials investigating HER3-DXd are currently underway, including TUXEDO-3 (patients with brain metastases and leptomeningeal seeding, NCT05865990), ICARUS-BREAST02 (patients who progressed after T-DXd treatment, NCT06298084), and SOLTI-2103 VALENTINE trial (patients with operable breast cancer, as neoadjuvant setting, NCT05569811).
Regarding safety outcomes, gastrointestinal and hematologic toxicities were the most common TEAEs (Supplemental Table 4). Grade ⩾3 TEAEs occurred in 71.4% of patients, including neutropenia (39.5%), thrombocytopenia (30.8%), anemia (18.6%), increased AST/ALT (6.0% each), and diarrhea (3.8%) in phase I/II trial (U31402-A-J101 study), in all doses. Hematologic AEs were managed through dose delays or reductions and did not lead to treatment discontinuation. The treatment discontinuation rate due to TEAEs is comparatively low (9.9%). 138
Notably, ILD/pneumonitis occurred in 6.6% of patients, with most cases presenting as grade 1 or 2. Three grade 3 and one grade 5 events were observed; the cause of death was deemed unrelated to ILD. The median onset time for treatment-related ILD was 141.5 days, ranging from 36 to 584 days. 138 In the ICARUS-BREAST01 trial, the incidence of treatment-related ILD was 7.1%, and all cases were grade 1 (Supplemental Table 4). 27 No patients in the SOLTI-TOT trial experienced HER3-DXd-related ILD/pneumonitis, although they received only a single neoadjuvant dose of HER3-DXd.139,140 Overall, TEAEs in patients receiving HER3-DXd are considered manageable through mitigation strategies, as previously discussed.
Enfortumab vedotin
Nectin-4 is a cell-adhesion molecule that is highly expressed in various types of cancers, including urothelial cell carcinoma, ovarian cancer, pancreatic cancer, lung cancer, and breast cancer.141–146 Its overexpression has been associated with tumor cell growth, angiogenesis, metastasis, and poor prognosis, particularly in breast cancer.147–149 EV is an ADC composed of mAb targeting nectin-4, a payload of MMAE, and a protease-cleavable linker. 150 The FDA has approved EV for treating patients with locally advanced or metastatic urothelial cell carcinoma. 151 For the patients with breast cancer, the EV-202 trial recently reported a confirmed ORR of 15.6% for the hormone receptor-positive/HER2-negative cohort (n = 45) and 19.0% for the TNBC cohort (n = 42) among those with locally advanced or mBC who had previously treated with cytotoxic chemotherapy. The median PFS rates for the hormone receptor-positive/HER2-negative and TNBC cohorts were 5.39 and 3.52 months, respectively. While EV demonstrated antitumor activity in both cohorts, the EV-202 trial did not achieve its prespecified ORR efficacy threshold. 113
In the EV-202 trial, the most frequently reported AEs in the hormone receptor-positive/HER2-negative cohort were fatigue (44.4%), pruritus (42.2%), maculopapular rash (40.0%), nausea (37.8%), dysgeusia (22.2%), diarrhea (22.2%), and peripheral sensory neuropathy (15.6%). The most common grade ⩾3 AEs were maculopapular rash (15.6%), pruritus (6.7%), and elevated AST levels (6.7%). Similarly, among patients with TNBC, the most frequent AEs included nausea (38.1%), pruritus (33.3%), maculopapular rash (31.0%), alopecia (28.6%), fatigue (28.6%), and dry skin (28.6%) 113 (Table 4). Notable AEs associated with EV therapy included skin reactions, peripheral neuropathy, dysgeusia, and hyperglycemia.
Skin toxicity
Skin reactions are attributed to the widespread expression of nectin-4 in the skin, representing on-target, off-tumor toxicity. 152 In the EV-202 trial, skin reactions occurred in 62.2% and 59.5% of patients within the hormone receptor-positive/HER2-negative and TNBC cohorts, respectively. Skin toxicities associated with EV typically present as erythematous, scaly, and pruritic papules, primarily affecting intertriginous, flexural, acral, and truncal regions while generally sparing the face. 153 Severe cutaneous AEs, including Stevens–Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN), are rare but reported in clinical trials of EV and highlighted in the US FDA label’s boxed warning. 151
The median time to onset of severe skin reactions following EV monotherapy administration was 0.6 months (range: 0.1–8 months). 146 Consequently, patients require close and frequent monitoring for cutaneous toxicity, especially during the first treatment cycle and throughout the duration of therapy.151,153 Prophylactic use of skin barrier protectants or sunscreen may be beneficial. Moreover, patient and caregiver education regarding the risk of severe skin reactions is crucial. 153 Grade 1 or 2 skin toxicities are often manageable with supportive care, such as topical corticosteroids combined with topical antibiotics and oral antihistamines. For grade 3 AEs, EV administration should cease until skin toxicity resolves to grade ⩽1. Treatment may then resume at the same dose level or be reduced by one dose level. 151 Oral corticosteroids may be administered to manage grade 3 AEs in addition to supportive care. If SJS or TEN is suspected—indicated by symptoms such as malaise, fever, mucosal involvement of the eyes, mouth, or genital area, or skin sensations of pain, burning, numbness, or tingling in the skin—EV should be discontinued immediately, and a dermatologist should be consulted for specialized management.153,154 In cases of confirmed SJS or TEN, or any grade 4 AEs or recurrent grade 3 AEs, EV should be permanently discontinued. 151
Hyperglycemia
In the EV-202 trial, any grade hyperglycemia occurred in 11.1% and 4.8% of the hormone receptor-positive/HER2-negative and TNBC cohorts, respectively. 113 In clinical trials evaluating EV monotherapy, any grade and grade ⩾3 hyperglycemia occurred in 17% and 7% of patients, respectively, with two reported fatalities. 151 Baseline hyperglycemia (38.2% vs 5.8%) and body mass index greater than 30 kg/m2 (29.3% vs 9.0%) were associated with a higher incidence of hyperglycemia. 155
The mechanisms underlying EV-associated hyperglycemia are poorly understood. 155 Baseline hemoglobin A1c should be assessed before initiating EV, and routine monitoring of non-fasting blood glucose levels is recommended prior to each treatment cycle. In cases of hyperglycemia, all potential etiologies, including corticosteroid use and infections, should be investigated. 156 If non-fasting blood glucose exceeds 250 mg/dL, EV should be held until glucose levels fall below 250 mg/dL, at which point EV can be resumed at the original dosage. 151
Peripheral neuropathy
Peripheral neuropathy associated with EV is attributed to the off-target uptake of the payload MMAE by peripheral nerves, disrupting microtubules and causing neurodegeneration.157,158 In the EV-202 trial, any grade peripheral neuropathy was reported in 26.7% of the hormone receptor-positive/HER2-negative cohort and 26.2% of the TNBC cohort. 113 In the EV-301 trial evaluating EV in patients with urothelial cell carcinoma, peripheral neuropathy was the most frequent reason for dose reduction (7.1%), dose interruption (15.5%), or withdrawal of treatment (2.4%). 155 Among patients receiving EV monotherapy in clinical trials, 89% experienced residual neuropathy, with 50% exhibiting grade ⩾2 neuropathy at their final evaluation. These findings indicate that peripheral neuropathy may be a persistent long-term consequence of EV treatment.
Patients should be monitored for new or worsening peripheral neuropathy, and treatment should be adjusted by interrupting or reducing the EV dose based on symptom severity. For patients experiencing grade ⩾3 AEs, permanent discontinuation of EV is recommended. 151 Adjunctive medications, including duloxetine, acetyl-L-carnitine, pregabalin, gabapentin, and mecobalamin, may benefit some patients with painful peripheral sensory neuropathy.156,158,159
Ladiratuzumab vedotin
LIV-1, a transmembrane protein involved in zinc transport, 160 has demonstrated an association between its expression and malignant progression and metastasis.161,162 Ladiratuzumab vedotin (SGN-LIV1A or LV) is an ADC comprising a LIV-1 targeting mAb, MMAE (payload), and a proteolytically cleavable linker. 163 The preliminary results in the phase I SGNLVA-001 trial (3 weekly schedules) demonstrated an ORR of 32% and median PFS of 11.3 weeks in a heavily pre-treated TNBC cohort. 164 The SGNLVA-002 trial (KEYNOTE-721, NCT03310957) and Morpheus-panBC trial (NCT03424005) are currently ongoing to assess the combination of LV and immune-checkpoint inhibitors pembrolizumab and atezolizumab, respectively.
Most AEs in the SGNLVA-001 trial were grades 1 or 2, with grade ⩾3 AEs comprising neutropenia (25%) and anemia (15%). Two cases of febrile neutropenia and one death due to sepsis were reported. 164 The safety profiles were similar in the weekly schedule, as the most common grade ⩾3 AEs were neutropenia (21%) and fatigue (14%; Table 4). 112 Detailed information on safety profiles has not yet been reported.
Disitamab vedotin
Disitamab vedotin (DV, RC-48) is a HER2-targeting ADC composed of mAb hertuzumab and MMAE payload, via a protease-cleavable linker. 165 DV was approved in China for treating HER2-positive locally advanced or metastatic gastric cancer. 166 DV is currently being assessed in clinical trials for the treatment of gastric cancer, urothelial cancer, biliary tract cancer, non-small-cell lung cancer, and breast cancer. 166 Recently, a phase II single-arm clinical trial in patients with breast cancer with PI3k/Akt/mTOR pathway activation demonstrated an ORR of 34.4% (9 of 26 patients) in those with HER2-positive mBC and 34.3% (12 of 35 patients) in those with HER2-low mBC. Patients with HER2-positive mBC experienced a median PFS of 4.5 months (95% CI 2.9–6.1), while those with HER2-low mBC had a median PFS of 3.4 months (95% CI 2.8–4.0). The most common grade ⩾3 AEs included neuropathy (11.3%), neutropenia (8.1%), elevated AST levels (3.2%), and leukopenia (3.2%); no treatment-related deaths were reported (Table 4). 114 Similar AEs were observed in phase II clinical trials involving patients with urothelial cell carcinoma, with the most common TRAEs were peripheral sensory neuropathy (68.2%), leukopenia (50.5%), elevated AST (42.1%), and ALT (35.5%). 167 These AEs were predominantly linked to MMAE payload, and their management strategies resemble those applied to other ADCs. 168
Sacituzumab tirumotecan
Sacituzumab tirumotecan (sac-TMT, SKB264/MK-2870) is a TROP-2-targeting ADC, which comprises mAb sacituzumab and belotecan-derivative topoisomerase I inhibitor, and a sulfonyl pyrimidine-CL2A-carbonate linker. 169 The payload exerts its antitumor activity by inducing cell cycle arrest at the G2/S phase following internalization into tumor cells. The pH-sensitive, hydrolytically cleavable linker facilitates the bystander effect in the extracellular environment. 169 Several clinical trials are evaluating sac-TMT in patients with gastric cancer, gastroesophageal junction cancer, endometrial cancer, non-small-cell lung cancer, and breast cancer.115,170–172
The phase III OptiTROP-Breast01 trial enrolled patients with locally advanced or metastatic TNBC who had received two or more prior chemotherapy regimens. Compared with physician’s choice chemotherapy (eribulin, vinorelbine, capecitabine, or gemcitabine), the sac-TMT arm demonstrated superior PFS (median PFS 6.7 vs 2.5 months, HR 0.32 (95% CI 0.22–0.44), p < 0.00001) and OS (median OS, not reached vs 9.4 months, HR 0.53 (95% CI 0.36–0.78), p = 0.0005). 115 The results of the exploratory analysis demonstrated that the sac-TMT arm achieved better clinical outcomes compared to the control arm regardless of prior PD-(L)1 therapy. 173 Based on the results of the OptiTROP-Breast01 trial, sac-TMT was recently approved for patients with metastatic TNBC who have received at least two prior systemic treatments in China. 174
The intervention and control arms demonstrated similar safety profiles. Patients in the sac-TMT arm experienced grade ⩾3 TRAEs and serious TRAEs at rates of 57.7% and 20.8%, respectively. The most common grade ⩾3 TRAEs were hematologic toxicities, including neutropenia (32%), anemia (28%), leukopenia (25%), and thrombocytopenia (9%). Furthermore, the incidence of peripheral neuropathy and ILD was low; only two patients (1.5%) experienced grade 1 peripheral neuropathy, and one patient experienced grade 2 ILD (Table 4). The reported safety profiles were consistent across clinical studies involving sac-TMT.172,175 Detailed safety information has not yet been published.
Arx788
ARX788 is a HER2-targeting ADC composed of an anti-HER2 mAb with AS269 (a tubulin inhibitor) and a non-cleavable linker. Mechanistically, AS269 conjugates site specifically to the mAb via a synthetic amino acid (para-acetyl phenylalanine), forming a highly stable oxime bone. This design improves the stability of ARX788 in systemic circulation and reduces off-target payload toxicity.124,176 ARX788 is currently under investigation for treating breast cancer, gastric cancer, gastroesophageal junction cancer, and other solid tumors.124,177,178
In the ACE-Breast02 trial, ARX788 demonstrated superior PFS compared with the lapatinib plus capecitabine arm (median PFS 11.33 vs 8.25 months; HR 0.64 (95% CI 0.49–0.82); p = 0.0006) in patients with HER2-positive breast cancer previously treated with trastuzumab and a taxane. 111 These results support the clinical efficacy of ARX788 as a potential subsequent treatment option for previously treated HER2-positive mBC. The ongoing ACE-Breast03 trial (NCT04829604) is evaluating the effectiveness of ARX788 in patients previously treated with T-DXd, potentially providing valuable insights into subsequent treatment options following T-DXd therapy.
The most frequently reported AEs included hepatotoxicity, ocular toxicity, alopecia, ILD, hypokalemia, hematologic toxicity, and gastrointestinal toxicity (Table 4). AE of special interest for ARX788 includes ILD and ocular toxicities. In the ACE-Breast02 trial, any grade and grade ⩾3 ILD were reported in 32.7% and 5.9% of patients, respectively. Three cases (1.4%) resulted in death. While the mechanisms of ARX-induced ILD are not fully elucidated, HER2-targeting mAbs, such as T-DXd and other HER2-targeting ADCs, may contribute to its development. Ocular toxicities manifested in 74.5% of patients enrolled in the ACE-Breast02 trial, primarily exhibiting as grade 1 or 2 (55.5%) with no occurrences of grade 4 or 5. The most common grade ⩾3 ocular events were blurred vision (12.3%), dry eye (9.1%), keratopathy (5.9%), and conjunctivitis (2.3%). 111 Ocular toxicities are likely attributable to the AS269 anti-tubulin payload, as similar toxicities are more frequently observed in ADCs containing anti-tubulin payloads, such as MMAF or DM4. 124 Management strategies for these AEs are similar to those employed for other ADCs.
Future directions
The toxicities of current ADCs primarily stem from cytotoxic payload at off-target sites and unwanted bystander effects. Understanding and managing the toxicities of ADCs are crucial for the continued development of these agents and their successful clinical application. To mitigate these toxicities, further research is needed to better elucidate the underlying mechanisms of specific toxicities, such as ILD and ocular toxicity. Furthermore, ADCs sharing the same antibody or payload can exhibit distinct toxicity profiles, necessitating further investigation into the mechanisms of ADC-related toxicity. For example, stomatitis is a common AE in patients receiving Dato-DXd but not in those receiving T-DXd, despite both utilizing the same payload (deruxtecan).
In addition, clinical trials, such as PRIMED, should investigate mitigation strategies for common toxicities like neutropenia and diarrhea in patients receiving SG. Future research should prioritize engineering highly specific mAbs for target antigens, developing substantially safer payloads, and designing novel linker technologies to mitigate off-target cleavage.
Novel ADCs targeting immune cells or the tumor microenvironment (TME), rather than tumor-associated antigens directly, are currently under development to improve cancer immunotherapy. For example, these novel immunostimulatory antibody conjugates (ISACs) utilize pattern recognition receptors (PRRs), such as Toll-like receptor 7/8 or stimulators of interferon genes agonists, instead of cytotoxic payloads. 179 Several ISACs employing PRR agonists with different targets, including HER2 and carcinoembryonic antigen, have already undergone early clinical trials.180–182 Several ADCs targeting specific components of the TME, such as T lymphocytes and fibroblasts, are currently under development.183–185 The toxicities associated with these novel ADCs are expected to elicit different mechanisms and presentations compared to current ADCs that utilize cytotoxic payloads. Therefore, a deeper understanding of the toxicities associated with these ADCs is crucial as the development of new ADCs continues.
Finally, personalized approaches that account for patient-specific factors and genetic predispositions are essential for mitigating toxicity. As described above, UGT1A1 is important in determining susceptibility to SG-induced toxicities, such as diarrhea and neutropenia. Furthermore, the current understanding of predisposing and significant risk factors for specific AEs, such as ILD associated with T-DXd, remains limited and requires further investigation. Consequently, additional research is necessary to assess patient-specific characteristics and genetic factors to enhance the tolerability of ADC therapy.
Conclusion
The advent of novel ADCs has significantly transformed the breast cancer treatment landscape. Given the rapid expansion of their clinical indications, a comprehensive understanding of associated AEs and effective management strategies, as well as proactive efforts to prevent and mitigate ADC-related toxicities, is essential. These toxicities predominantly result from releasing the cytotoxic payload at unintended sites, causing undesirable off-target toxicities. Furthermore, mAbs and linker components of ADCs contribute to their overall toxicity profile. However, several mechanisms driving ADC-related toxicities remain unclear, highlighting the need for additional research. Understanding the mechanisms driving these toxicities is essential for designing safer ADC candidates and developing optimal patient management strategies.
Management strategies, including dose reduction and treatment delays, can mitigate several severe adverse effects, such as hematologic and gastrointestinal toxicities. Early screening, suspicion, and prompt identification of significant AEs, such as ILD and pneumonitis, are crucial for improving patient outcomes. Education for both the multidisciplinary healthcare team and patients is essential for ensuring prompt and timely management of AEs. Furthermore, consultations with subspecialists, such as dermatologists and ophthalmologists, are essential for managing organ-specific AEs and optimizing patient care. Moreover, careful assessment of the potential for synergistic or overlapping toxicities is essential when combining ADCs with other anticancer therapies. Proactive management and interdisciplinary collaboration to address these challenges will enhance the safety and efficacy of ADCs in breast cancer treatment, ultimately improving clinical outcomes.
Supplemental Material
sj-docx-1-tam-10.1177_17588359251324889 – Supplemental material for Toxicities and management strategies of emerging antibody–drug conjugates in breast cancer
Supplemental material, sj-docx-1-tam-10.1177_17588359251324889 for Toxicities and management strategies of emerging antibody–drug conjugates in breast cancer by Sora Kang and Sung-Bae Kim in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.