
Abstract
Background:
Despite advances in the treatment of metastatic castration-resistant prostate cancer (mCRPC), primary and secondary resistance to current therapies remains. Elevated circulating sphingolipids are associated with poor outcomes in patients with mCRPC, including therapeutic resistance and shorter overall survival. PCPro is a clinically accessible, regulatory compliant plasma lipid biomarker of poor prognosis in mCRPC, which incorporates prognostic sphingolipids. We hypothesize that reversal of the PCPro signature in men with mCRPC by sphingolipid-lowering agents will improve their clinical outcomes. However, the first step is to determine whether this poor prognostic lipid signature can be modulated. A potential sphingolipid-lowering agent is the PCSK9-inhibitor evolocumab, which is used in the management of hypercholesterolemia.
Objectives:
Our primary objective is to assess whether treatment with evolocumab during standard anticancer therapy can safely modify the PCPro signature in men with mCRPC.
Design:
This is a multicenter, open label phase II trial.
Methods:
Men with mCRPC commencing docetaxel, cabazitaxel, abiraterone, enzalutamide, olaparib, or lutetium-177 PSMA for disease progression will be screened for the presence of PCPro. Those who are PCPro positive will receive a 12-week course of evolocumab concurrent with their standard therapy. Dosage is as per cardiovascular guidelines (420 mg subcutaneously every 4 weeks). PCPro will be repeated after 12 weeks. The primary endpoint is reversal of PCPro. The secondary endpoint is the safety of combination therapy with exploratory endpoints characterizing changes in comprehensive lipid profiles pre- and post-treatment.
Discussion:
This study will evaluate whether evolocumab can safely modify the PCPro signature in men with mCRPC, providing essential data to the development of precision metabolic therapy in the management of prostate cancer.
Trial registration:
This study is approved by the Human Research Ethics Committee (X22-0072 and 2022/ETH00427). It is registered with the Australian New Zealand Clinical Trials Registry (ACTRN12622001003763).
Keywords
Background
Prostate cancer (PC) is the second most commonly diagnosed cancer in men worldwide. 1 While it is often diagnosed at a localized and potentially curable stage, over 375,000 men die from advanced PC globally each year. 1 Despite advances in the management of metastatic PC over the last decade, drug resistance remains the key cause of lethal disease. 2 The major oncogenic driver for PC is androgen receptor (AR) signaling. Androgen deprivation therapy (ADT) is therefore the cornerstone of treatment. At first diagnosis, PC is referred to as hormone-sensitive (HSPC) due to the high response rates to ADT. Over time, PC becomes resistant to ADT and is referred to as castration-resistant (CRPC), the lethal form of PC. AR pathway inhibitors (ARPIs), taxanes, poly-ADP ribose polymerase inhibitors (PARPi), and targeted radioisotopes (e.g., lutetium-177-PSMA-617) improve survival and quality of life in men with advanced PC. 3 However, in the case of each of these drugs a proportion of men have innate resistance to individual therapies, and all acquire resistance over time. 3 Longer-term control of advanced PC requires approaches that address the multiple hallmarks of cancer, incorporating the cancer genome, immune system, and metabolism, which all contribute to cancer progression and treatment resistance.4–6
Molecular and animal studies have demonstrated that lipid metabolism is dysregulated in PC. 7 Plasma lipidomic profiling studies of mCRPC have established that certain circulating sphingolipids, in particular ceramides, are associated with shorter progression-free survival and overall survival (OS).8–10 Elevated ceramides are also associated with increased risk of metastatic relapse in localized disease and earlier treatment resistance in mHSPC.8,9 In noncancer patients, elevated circulating sphingolipids are observed in those at increased risk of cardiovascular events.11,12 Plasma concentrations of these sphingolipids can be modified with statins 13 and proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors 14 resulting in a decreased incidence of cardiovascular events. PCSK9 inhibitors are potent cholesterol-lowering agents that are used to manage hypercholesterolemia in cases where statins alone are insufficient. 15 PCSK9 binds to the low-density lipoprotein-receptor (LDL-R) on the surface of hepatocytes, leading to its degradation. Inhibition of PCSK9 therefore inhibits degradation of the LDL-R, thus increasing LDL-R mediated removal of LDLs from the circulation. In addition to dramatically lowering LDL-cholesterol, 16 PCSK9 inhibitors lower the circulating levels of sphingolipids, and their role in the modulation of the wider lipidome is increasingly recognized. 14 There is also increasing evidence of aberrant PCSK9 expression in cancer in general,17,18 and PCSK9-inhibitors may enhance anticancer therapy via both lipid and nonlipid mediated pathways.18,19
We have developed a plasma lipid biomarker assay named PCPro, which incorporates prognostic sphingolipids and is able to prospectively identify men with who are more likely to be abiraterone or enzalutamide-resistant (median OS 31.5 months in men treated with an ARPI who were PCPro negative at baseline vs 12.3 months in men who were PCPro positive; hazard ratio (HR) 2.36, 95%CI 1.47–3.80, p < 0.001). PCPro-positive men with mCRPC have significantly shorter survival overall than those who are PCPro-negative (HR 3.75, 95% CI 2.29–6.15, p < 0.001 in the discovery cohort and HR 2.13, 95%CI 1.46–3.12, p < 0.001 in the validation cohort) 20 (Figure 1). PCPro was developed in accordance with National Pathology Accreditation Advisory Council (NPAAC) guidelines and has been validated across two independent cohorts of 288 men. 20 PCPro is subject to a provisional patent (PCT/AU2023/050849).
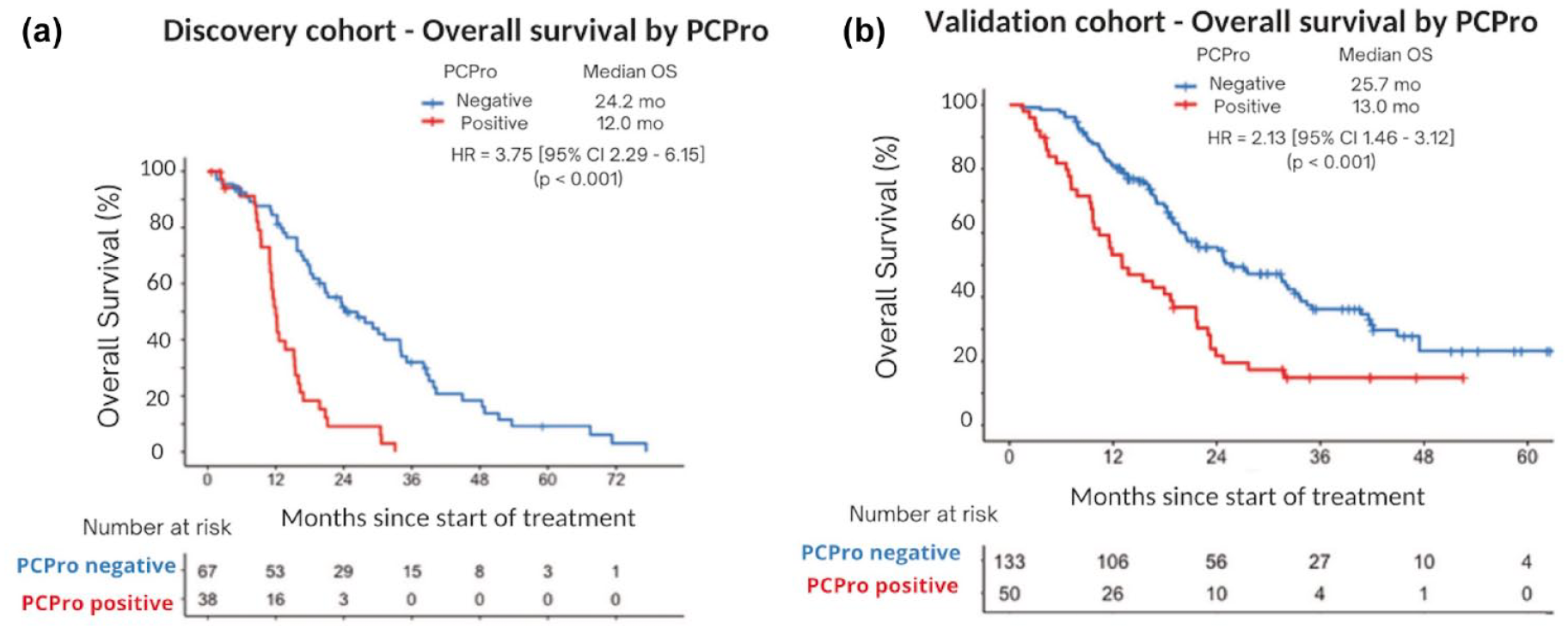
Kaplan–Meier survival analysis of overall survival by PCPro in (a) discovery and (b) validation cohorts. This figure has been reproduced and was originally published in Prostate Cancer and Prostatic Diseases (Scheinberg et al, 2023), 20 under a Creative Commons Attribution 4.0 International License.
This proof-of-concept, phase II, single-arm clinical trial will aim to establish whether PCPro can be measured prospectively in a clinical trial, and whether it is modulated by lipid-targeted therapy. This is the first study to use PCPro to prospectively select patients for lipid-targeted therapy in a clinical trial setting. Specifically, the trial will evaluate whether the PCSK9 inhibitor, evolocumab, can modulate circulating ceramide concentrations in PCPro positive-men and alter the poor prognostic lipid signature.
Study objectives
The primary objective of this phase II clinical trial is to evaluate whether treatment with the PCSK9-inhibitor, evolocumab, during standard anticancer therapy can modify the poor prognostic lipid signature (PCPro) in men with mCRPC. Standard anticancer therapies in this context include chemotherapy (docetaxel or cabazitaxel), an androgen-receptor signaling inhibitor (abiraterone or enzalutamide), lutetium-177 PSMA therapy, or PARPi therapy (olaparib).
The secondary objective is to determine whether treatment with evolocumab concurrently with standard anticancer therapy affects the incidence of adverse events. Finally, an exploratory biomarker study will be performed on plasma samples taken at baseline and following completion of 12 weeks of evolocumab therapy. This will involve comprehensive lipidomic profiling of pre- and posttreatment plasma samples, allowing a broader analysis of changes in circulating lipids with evolocumab treatment.
Trial design
This trial is a multicentre, open label phase II study of evolocumab in addition to chemotherapy (docetaxel or cabazitaxel), ARPI therapy (abiraterone or enzalutamide), PARPi therapy (olaparib), or lutetium-177 PSMA therapy in patients with mCRPC. In this trial, men with mCRPC starting a new anticancer therapy (any line) for disease progression will be screened for the presence of the PCPro lipid signature. Those who are PCPro positive will receive a 12-week course of evolocumab concurrent with their standard anticancer therapy. Dosage is as per hypercholesterolaemia guidelines at 420 mg subcutaneously every 4 weeks. PCPro will be repeated after 12 weeks (see study schema in Figure 2).

Study schema.
Participant population
The target population is adult men with mCRPC commencing docetaxel, cabazitaxel, abiraterone, enzalutamide, olaparib, or lutetium-177 PSMA therapy for disease progression. Men will be pre-screened for the presence of the poor prognostic lipidomic profile. Only those who are PCPro positive will receive evolocumab. This study will be conducted at three cancer centers in New South Wales, Australia. See Table 1 for full inclusion and exclusion criteria.
Key inclusion and exclusion criteria.

ALP: alkaline phosphatase; ALT: alanine aminotransferase; AST: aspartate aminotransferase; ECOG: European Cooperative Oncology Group; GFR: Glomerular filtration rate; mCRPC: metastatic castration resistant prostate cancer; PCWG3: Prostate Cancer Clinical Trials Working Group 3; ULN: upper limit of normal.
Intervention
Evolocumab is a fully humanized monoclonal antibody that binds to PCSK9, promoting the degradation of this enzyme. The recommended dose of evolocumab to treat hypercholesterolaemia in cardiovascular disease is either 420 mg every 4 weeks (administered as a subcutaneous injection), or 140 mg every 2 weeks.16,21 No dose adjustment is required in patients with chronic kidney disease or mild-to-moderate hepatic impairment. It has not been studied in severe hepatic impairment. Evolocumab achieves significant reduction in circulating lipids within the first 12 weeks of therapy. 16
In a large pooled analysis of 12 trials evaluating evolocumab, the pooled estimate for any treatment-related adverse event at 12 weeks follow-up was 53%, which was not significantly different from placebo (45%, relative risk (RR) 1.07, 95%CI, 0.95–1.21). 16 Adverse events were generally mild to moderate. Serious treatment related adverse events occurred in 1.9% of patients, and adverse events leading to discontinuation occurred in 1.6%. At 52 weeks follow-up, the most common adverse events (of any grade) in the evolocumab arm included nasopharyngitis (11.5%), musculoskeletal and connective tissue disorders (9.2%), and upper respiratory tract infection (8.5%) or influenza (7.3%). 16
In this study, patients will be given evolocumab 420 mg via subcutaneous injection every 4 weeks, for 12 weeks, commencing on day one of the first dose of chemotherapy, ARPI therapy, PARPi therapy, or lutetium-177 PSMA therapy (i.e., three 4-weekly doses will be administered). Dose modifications are not permitted. Patients who need to come off the study drug for any reason (including adverse events or participant request) will be discontinued. Participants who discontinue the study drug will continue follow-up safety assessments as outlined below. For participants who cannot attend or decline follow-up visits, information can be obtained by phone. This protocol (version 2.5 dated April 10, 2024) completely adheres to the Standard Protocol Items: Recommendations for Interventional Trials statement, 22 see Supplementary File 1.
Assessments
Plasma collection
Blood for plasma lipidomic studies will be taken at baseline (pretreatment). Men who are PCPro positive and proceed to evolocumab treatment will have another blood sample taken at 12 weeks (posttreatment). Blood samples are nonfasting and will be collected in BD Vacutainer tubes containing K2EDTA and processed by centrifugation, initially at 1600g for 15 min at room temperature, and then at 5000g for 10 mins. The plasma will then be removed by transfer pipette and aliquoted into cryovials for storage at −80°C.
Hemoglobin A1C (HbA1c) will also be determined at baseline and posttreatment. In addition, patients will undergo routine blood tests every 3–6 weeks as part of their standard anticancer therapy. The blood tests will include electrolytes, a full blood count, renal function, liver function, and prostate-specific antigen.
Lipidomic profiling
Patient plasma will be analyzed at baseline for PCPro biomarker status. This method has been described previously. 20 In summary, the PCPro biomarker assay produces a risk score derived from the plasma levels of Cer(d18:1/18:0), Cer(d18:1/24:0), Cer(d18:1/24:1), total cholesterol, and total triglycerides. The risk score is calculated as follows:

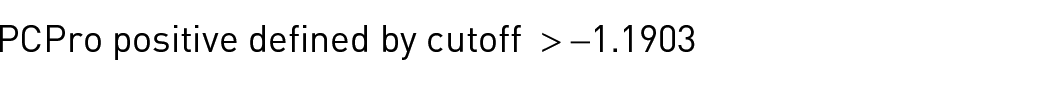
A high-risk score (greater than −1.1903) is defined as ‘PCPro positive’ and is associated with poorer prognosis. Plasma levels of ceramides are determined by a targeted Liquid Chromatography-Mass Spectrometry (LC-MS) assay. Total cholesterol and triglycerides are determined by standard enzymatic colorimetric assays using the COBAS 80000 analyzer (module C702) (Roche). Men who are PCPro positive at baseline and go on to complete 12 weeks of evolocumab treatment will then have their PCPro status reanalyzed in a posttreatment blood test. In addition, baseline and posttreatment plasma samples will undergo high-throughput lipidomic analysis of >800 lipid species with relative quantitation using LC-MS as described previously. 9
Safety assessments
Safety assessments will be performed prior to each dose of evolocumab and again at 21–24 days after completion of the last dose. For patients on chemotherapy (docetaxel or cabazitaxel), assessments during treatment will occur every 3 weeks as per standard practice. For patients on ARPI therapy (abiraterone or enzalutamide), PARPi therapy (olaparib), or lutetium-177 PSMA therapy, assessments will occur every 4 weeks. Assessments during treatment may be performed within 7 days of the specified time point, unless otherwise specified. If dose delay or cessation of docetaxel, cabazitaxel, abiraterone, enzalutamide, olaparib, or lutetium-177 PSMA therapy occurs for any reason on the study, treatment with evolocumab will continue until completion after 12 weeks.
Study endpoints
The primary endpoint is change in the poor prognostic lipid signature, PCPro, following treatment with evolocumab. The main secondary endpoint is treatment toxicity of the combination of standard anticancer therapy with evolocumab, which will be measured by the rate of adverse events assessed at each clinic visit. These will be graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (CTCAE) version 5.0 (National Institutes of Health, United States of America). As an exploratory endpoint changes in the concentrations of >800 lipid species following treatment with evolocumab will also be determined.
Statistical considerations
The planned sample size of this single-arm phase II study is 30 patients. A sample size of thirty men, who are PCPro positive, will provide 80% power, with a type 1 error of 10%, to distinguish a PCPro conversion rate of >50% versus <30%. Based on our previous data, 27%–35% of men with mCRPC will be PCPro positive at baseline. We therefore aim to screen 120 men. An interim analysis will be performed after the first 10 patients who are PCPro positive at baseline have completed 12 weeks of evolocumab therapy. The study will be stopped if two or less of these participants are PCPro negative at the end of treatment (indicating a PCPro conversion rate of < 20%). Only participants who complete the full course of evolcoumab therapy and have both baseline and end-of-treatment plasma samples available for assessment will be included in the analysis to evaluate whether evolcoumab can modulate PCPro. Any participant who starts evolocumab, regardless of completion, will be included in the evaluation of adverse events.
Statistical analyses will be performed with the software R, Microsoft Excel, and/or other appropriate statistical software. PCPro status will be calculated using the formula outlined above. When evaluating the larger dataset of > 800 lipid species, differences in lipid levels at baseline versus posttreatment will be assessed by paired sample t-tests. p-Values less than 0.05 will be considered statistically significant.
Initial experience with use of PCPro in a prospective clinical trial
Patient enrolment for this study commenced in March 2023 at three sites in NSW, Australia. The anticipated study completion date is December 2025. To date, 34 patients have been screened, of which nine are PCPro positive, representing a positive rate of 26.5%. This is the first time PCPro has been used to direct therapy, so the initial stage of this trial involved ensuring that PCPro can be run in a timely manner in a clinical reference laboratory. The trial specified a 2-week turnaround between collecting the initial blood sample and dissemination of the PCPro result. This time frame was met for 33/34 (97%) patients screened, with a median of 8 days (range 1–18) between blood sampling and result dissemination. The one patient for which result dissemination fell out of the 14-day window (at 18 days) was due to unplanned staff leave. Further staff have now been trained on the laboratory method.
This is also the first time that the PCPro assay is being performed on one to two patient samples at a time, rather than in larger batches (as performed during method development). Part of the initial stage of this trial therefore involved ensuring reliable outputs are maintained for single sample analysis. This was assessed after the first five patients had been screened. All five patient samples were run initially as individual assays (as they were screened on different days over a 117-day period). They were then rerun in one batch. For this batch analysis, plasma was thawed and a fresh 100 µL was extracted from each patient sample according to the standard protocol, and analyzed on the LC-MS. The remaining plasma was refrozen at −80°C. The percent difference in concentration of each analyte in the batch analysis compared with the single assays was determined. The mean percent difference for each analyte was calculated from the results of all five plasma samples, and was < 6.6% for each analyte (Table 2). This is within the expected measurement uncertainty (MU) for each analyte based on the initial validation of PCPro 20 (where MU is 2 × the coefficient of variation for each analyte, giving a corresponding level of confidence of 95%).
Mean percent difference between batch analysis and single assays for each of the three analytes of interest (across five patient samples).

Conclusion
Innate and acquired treatment resistance is an ongoing issue for mCRPC. The need for new treatments that can overcome resistance to improve quality of life and survival for men with mCRPC is vital. Dysregulated lipid metabolism is increasingly recognized as a resistance mechanism in metastatic PC and represents a potential target for lipid metabolic therapy. We have developed a validated, regulatory compliant lipid biomarker named PCPro that is able to prospectively identify men with mCRPC who are more likely to be resistant to ARPI therapy, and have shorter OS. The next step is to determine whether this prognostic lipid biomarker can be modulated. This trial will demonstrate whether 12 weeks of a PCSK9 inhibitor (evolocumab), concurrent with standard anticancer therapy, can modify the PCPro signature in men with mCRPC who are PCPro positive at baseline. This is a proof-of-concept clinical trial and is the first trial evaluating the use of PCPro as a biomarker to direct therapy. It will provide invaluable information toward the goal of precision metabolic therapy. It will also provide insights into the potential adverse effects and safety of combination lipid metabolic therapy with standard anticancer therapy to overcome treatment resistance. However, given this trial does not include a control arm and it allows for standard anticancer therapies with quite different toxicity profiles, it will be limited in its ability to analyze adverse events of combination therapy. In addition, this trial is not designed to determine whether a short course of lipid-lowering therapy can alter oncological outcomes. Should this trial be positive overall, this will be evaluated in a larger, randomized controlled trial.
Supplemental Material
sj-pdf-1-tam-10.1177_17588359241307814 – Supplemental material for Evolocumab in metastatic castration-resistant prostate cancer: study protocol for a single-arm, phase II trial, and initial experience with use of a validated lipid biomarker to direct therapy
Supplemental material, sj-pdf-1-tam-10.1177_17588359241307814 for Evolocumab in metastatic castration-resistant prostate cancer: study protocol for a single-arm, phase II trial, and initial experience with use of a validated lipid biomarker to direct therapy by Rhiannon Mellor, Luke Ardolino, Tahlia Scheinberg, Michael Fitzpatrick, Hui-Ming Lin, Paul Bonnitcha, David Sullivan, Peter J. Meikle, Martin R. Stockler, Tania Moujaber, Anthony Joshua and Lisa Horvath in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.