
Abstract
Background and objectives:
Breast cancer is the most common cancer in women, with one in eight women suffering from this disease in her lifetime. The implementation of centrally organized mammography screening for women between 50 and 69 years of age was a major step in the direction of early detection. However, the participation rate reaches approximately 50% of the eligible women, one reason being the painful compression of the breast, cited as a major issue for not participating in this very important program. Therefore, focusing current research on less painful and less invasive techniques for the detection of breast cancer is highly clinically relevant. Liquid biopsies offer this option by detection of distinct molecules such as microRNAs (miRNAs) or circulating tumor DNA (ctDNA) or disseminated tumor cells.
Design and methods:
Here, we present the first proof-of-concept approach for sequencing miRNAs in female urine to detect breast cancer and, subsequently, intrinsic subtype-specific miRNA patterns and implement in this regard a novel random forest algorithm. To this end, we performed miRNA sequencing on 82 urine samples, 32 samples from breast cancer patients (9× luminal A, 8× luminal B, 9× triple-negative, and 6× HER2) and 50 healthy control samples.
Results and conclusion:
Using a random forest algorithm, we identified a signature of 275 miRNAs that allows the detection of invasive breast cancer in urine. Furthermore, we identified distinct miRNA expression patterns for the major intrinsic subtypes of breast cancer, specifically luminal A, luminal B, HER2-enriched, and triple-negative breast cancer. This experimental approach specifically validates miRNA sequencing as a technique for breast cancer detection in urine samples and opens the door to a new, easy, and painless procedure for different breast cancer-related medical procedures such as screening but also treatment monitoring.
Plain language summary
Breast cancer is the most common cancer in women, with one in eight women getting it during their lives. To catch it early, a regular screening program using mammograms was set up for women aged 50 to 69. However, only about half of the eligible women participate, partly because mammograms can be painful due to the breast compression involved. Therefore, researchers are looking for less painful ways to detect breast cancer. One promising method is called a liquid biopsy, which looks for specific molecules like miRNAs or ctDNA in bodily fluids. In this study, we explored using urine samples to detect breast cancer by sequencing miRNAs. We analyzed urine from 82 women, including 32 with breast cancer and 50 healthy women. We used a special computer algorithm called a random forest analysis to identify a pattern of 275 miRNAs that could indicate the presence of breast cancer in urine. We also found specific miRNA patterns for different types of breast cancer: luminal A, luminal B, HER2-enriched, and triple-negative. This research shows that miRNA sequencing of urine samples can be a new, easy, and painless way to detect and monitor breast cancer, potentially improving screening and treatment monitoring.
Keywords
Background
Breast cancer (BC) is still the leading cause of cancer-related deaths among the female population, affecting one in eight women in her lifetime. 1 Moreover, this disease still places an enormous burden on healthcare systems worldwide. In addition to tumor biology, which guides treatment options and therapeutic needs in general, tumor stage is still an important risk factor for treatment decisions. Implementing mammography-based early detection programs consecutively led to decreased tumor size (T stage) and decreased axillary lymph node involvement (N stage), thereby improving survival rates in patients with BC. 2 Almost 100% of German women aged 50–69 years are invited to undergo mammography screening every 2 years. However, since its implementation in 2009, the participation rate has stagnated at approximately 50%. 3
Therefore, it appears reasonable that these programs could dramatically profit from new early detection methodologies, which are less invasive than the current standards.
MicroRNAs (miRNAs) are small noncoding RNA molecules that regulate gene expression by binding to specific target mRNAs, affecting their translation. They are found in many different cell types and tissues and have been shown to play a role in a wide range of biological processes, including cell growth and proliferation, differentiation, and apoptosis. Recent studies have shown that miRNAs can also be found in body fluids, such as blood and urine, 4 where they can be easily isolated and quantified and can act as biomarkers for various diseases, including cancer.5,6
As biomarkers, miRNAs possess high potential for cancer detection because they are stable in body fluids, which allows easy collection and storage of samples. 7 They are also present and detectable in very small samples, which makes them ideal biomarkers. 8 In addition, miRNA-based diagnostic tests can be noninvasive, which is especially important for the early detection of cancer.
While, several studies have investigated the use of miRNAs as biomarkers for BC and identified miR-21, miR-155, miR-205,9 –11 miR-424, and miR-42312,13 as potential biomarkers for this disease, additional research is needed to fully establish the clinical utility of miRNA-based diagnostic tests for BC. Generally, miRNAs regulate multiple processes in the body and are used to amplify or buffer cellular signaling programs. Therefore, it seems unlikely to identify reliable single marker miRNAs in body fluid from a heterogeneous population of female cancer patients. By contrast, it seems rather feasible to investigate changes in groups of miRNAs regulating processes, especially in the context of cancer.
To investigate the feasibility of whole miRNA genome detection as a diagnostic tool for BC, we investigated a cohort of 82 urine samples from BC patients and healthy individuals using miRNA sequencing for the first time. The goal was to identify all currently known miRNAs regulated in BC in a completely unbiased setting without prior selection of BC-specific miRNAs from the literature. In this proof of concept, we present the first step toward the clinical use of miRNA sequencing in BC detection and provide insight into the stratification of BC patients utilizing this information.
Methods
Patient selection and urine sample preparation
Samples were collected at the University Hospital Aachen (ethics vote 206/09) and University Hospital Erlangen. Participants in Erlangen were recruited within the iMODE-B study (Imaging and Molecular Detection of Breast Cancer; ethical approval by the ethics committee of the Friedrich-Alexander-Universität Erlangen-Nuremberg; #325_19 B). Patients were eligible for inclusion if they indicated a diagnostic biopsy due to a suspicious breast lesion. The main aim of the iMODE-B study was to identify molecular markers that are predictive of patient prognosis and treatment response at the time of the first diagnosis of BC. After the participants provided written informed consent in accordance with the Declaration of Helsinki, biospecimen sampling was performed.
A total of 355 urine samples were collected between November 2019 and July 2020. The urine samples were centrifuged at 944g for 10 min at room temperature to separate the cell pellet and cell-free supernatant, which were stored separately at −80°C until further use. For miRNA extraction, at least 7 ml of cell-free supernatant was available, 4 ml was used for miRNA extraction, and the remaining volume was used as a backup. From these 282 participants, 82 were randomly selected (n = 50 healthy controls and n = 32 cancer patients) for final analysis.
MiRNA sequencing and statistical analysis
Sequencing libraries were prepared with the QIASeq miRNA UDI Library Kit (Qiagen, Hilde, Germany) according to the manufacturer’s instructions. To the recommended 4 µl sample input for biofluids, 1 µl of synthetic miRNAs from the QIASeq miRNA Library QC Kit was added as additional quality control. The quality of the libraries was checked on a Bioanalyzer or Tapestation (both Agilent, Waldbronn, Germany), and the libraries were quantified by a Quantus fluorometer (Promega, Madison, WI, USA). All the samples were sequenced on an Illumina NextSeq 500 instrument (Illumina, San Diego, CA, USA) in 72 bp single-end mode. Sequencing yielded a mean coverage of approximately five million reads per sample.
FASTQ files were generated using bcl2fastq (Illumina). To facilitate reproducible analysis, samples were processed using the publicly available nf-core/smRNAseq pipeline version 2.2.1 14 implemented in Nextflow 23.04 15 using Docker 24.0.2 with minimal command. All analyses were performed using custom scripts in R version 4.2.2 using the DESeq2 v.1.38.3 framework. 16
One low-quality sample was excluded, leaving data from 32 cancer patients and 49 healthy individuals for downstream analyses. We normalized read counts using DESeq2 and employed a fivefold cross-validation approach to ensure robust evaluation. Due to the limited sample size, all samples were included in the training process.
Our analysis strategy involved two key steps: first, we applied a random forest (RF) algorithm to all 4039 miRNAs to explore broad interactions. Next, we used RF-based feature selection to identify the most relevant miRNAs. Supervised learning was conducted using a multi-label classification approach. Python tools such as scikit-learn, numpy, pandas, matplotlib, and seaborn were used for analysis and visualization, facilitating a comprehensive exploration of miRNA–target dynamics.
Quantitative PCR
RNA isolation was performed with the miRNeasy Mini Kit by Qiagen (#217004) following the instructions in the user manual. One 5 ml aliquot was isolated to ensure the same volume for each sample. After isolation, the RNA was stored at −80°C until cDNA transcription. Transcribed miRNA samples were analyzed using a TaqMan Advanced miRNA Assay (#A25576 Applied Biosystems, individual assays were purchased for each investigated miRNA, sequences are listed on Applied Biosystems Website) in combination with TaqMan Fast Advanced Master Mix (#4444557 Applied Biosystems, Darmstadt/Germany). A Roche LightCycler 480 Instrument II (#05015243001) was used for detection. The samples and master mix were added to 384-well plates (#04729749001 LightCycler 480 Multiwell Plate white; Roche, Grenzach-Whylen/Germany). For correct preparation following the manufacturer’s instructions, the samples were diluted with 0.1× TE. Samples were pipetted in triplicate on each plate per assay and the exogenous control ath-mir-159a. Primer sequences used are shown in Supplemental Table 2. The LightCycler 480 data were exported as MS-EXCEL files and analyzed. The resulting Ct values were analyzed using the ∆∆Ct method. The microRNA ath-mir-159a was used as a reference gene to normalize the data to the ΔCt, and samples from healthy donors served as the second reference to calculate the miRNA fold change.
GraphPad Prism software was used for statistical evaluation. Student’s t-test was used to determine significant differences.
Results
Analysis of 82 urine samples from BC patients and healthy female individuals
We implemented miRNA sequencing as an unbiased method to evaluate the currently known miRNA genome from human urine samples. The first aim was to implement reliable and consistent detection of miRNAs in urine. Since this study should evaluate the feasibility of miRNA sequencing from reasonably small sample sizes, which can be obtained during a regular visit in an outpatient setting, we tested a sample size of 4 ml of urine in this sequencing approach.
Eighty-two individual patient samples were utilized in this study, and the miRNAs were extracted from 4 ml urine samples as described in the Methods section. The 82 samples were classified into 50 healthy tumor-free control, 6 HER2-enriched, 9 luminal A, 8 luminal B, and 9 triple-negative breast cancer (TNBC) tumor-bearing patient urine samples (Table 1).
Description of the patient cohort used for analysis.

HER2, Her2-enriched; TNBC, triple-negative breast cancer.
MiRNA sequencing of urine samples enables the detection of more than 4000 target miRNAs
We analyzed the expression of more than 4600 miRNAs and detected the consistent expression of 4039 distinct miRNAs. The mean absolute miRNA expression (normalized read counts) of all the samples varied between 6.5 and 14.1, with the majority of the samples showing an average expression of 7–8 ± 1 (Supplemental Figure 1). In the first step, we attempted to verify a couple of regulated miRNAs by qPCR. Using a 1.5-fold log change upregulation or downregulation of expression with an adjusted p value of 0.05 or less as a cutoff, we identified several miRNAs exhibiting differential expression between healthy and tumor-bearing individuals (Figure 1(a) and Supplemental Table 1). Unfortunately, expression levels of most of these miRNAs identified in the Volcano-Blot Figure 1(a) were close to or below a reasonable level of detection for the qPCR-based analysis that we intended to establish. Nevertheless, we tried to further stratify patient subgroups and received a number of differentially highly expressed miRNAs for luminal A versus healthy (161 miRNAs), HER2 versus healthy (30 miRNAs), luminal B versus healthy (19 miRNAs), and TNBC versus healthy (12 miRNAs) patients. Several of these differentially expressed miRNAs were found in more than one of the subgroup comparisons and could therefore not be used as biomarkers to stratify individual patients.

Initial analysis and evaluation of the sequencing data. (a) Volcano plot showing a 1.5-fold log change upregulation or downregulation of expression with an adjusted p value of 0.05 or less as a cutoff (miRNAs identified as red dots were considered significant; those identified as not significant are shown in blue). (b) Subclasses of breast cancer could be identified by groups of miRNAs.
Confirmation of miRNA expression profiles by qPCR
To validate our initial results of the sequencing, we analyzed the expression of the most highly differentially expressed miRNA markers using qPCR and confirmed the differential regulation of some (Supplemental Figure 2(A)–(H)), but not others (Supplemental Figure 3(A)–(G)). Overall, it must be stated that the variability in the qPCR results far exceeded the variability found in the sequencing results. Nevertheless, some subgroup-specific expression patterns, such as high expression of miR-30a-5p in TNBC, were validated (Supplemental Figure 2(F)). In general, the detection of cancer in urine compared to urine from healthy individuals was more consistent even though it varied distinctly among the whole patient population.
The RF approach for data modeling
Even if the generic statistical approach that we employed to address the marker identification with a classical paradigm captured overall trends of miRNAs across samples, it nevertheless failed to detect combinatorial expression patterns of multiple miRNAs in a patient-specific manner. We did not identify a unique reliable marker in this manner; we hypothesized that some important miRNAs might not exhibit strong individual distinctions in tumors; instead, their collective expression pattern is decisive. Therefore, we applied machine learning methods capable of detecting these patient-specific combinatorial patterns and identifying potential biomarker groups of miRNAs.
An analysis based on the miRNA sequencing data showed that the unsupervised clustering of the whole dataset did not match distinct patterns within the cancer group or a subtype (Figure 2(a)). Moreover, principal component analysis (PCA) did not reveal any distinct patterns (Figure 2(b)). To investigate the data in greater detail, we applied several machine-learning algorithms to detect hidden patterns of miRNA expression. Figure 2(c) shows the initial benchmark of the shallow learning methods. The RF outperforms logistic regression, decision tree, and support vector machine (SVM) due to its ensemble approach, which reduces overfitting, captures complex relationships, handles high-dimensional data, and balances bias-variance trade-offs by aggregating diverse decision trees.

Data analysis. (a) Unsupervised clustering of the whole dataset of miRNA sequencing data. (b) PCA. (c) Analysis of several machine learning algorithms to detect hidden patterns of miRNA expression. The initial benchmarks of the learning methods used are as follows: logistic regression, decision tree, random forest, and SVM.
Sequencing data indicating the ability of 275 individual miRNAs to distinguish BC patients from healthy women
We trained the RF model with two approaches: one with 4039 miRNAs and the other with feature selection via RF (Figure 3). The prediction with 275 miRNAs selected by the RF algorithm (mean area under the curve (AUC) = 0.67; Figure 3(a), right) performed much better than the prediction with the whole miRNA dataset (mean AUC = 0.58; Figure 3(a), left).
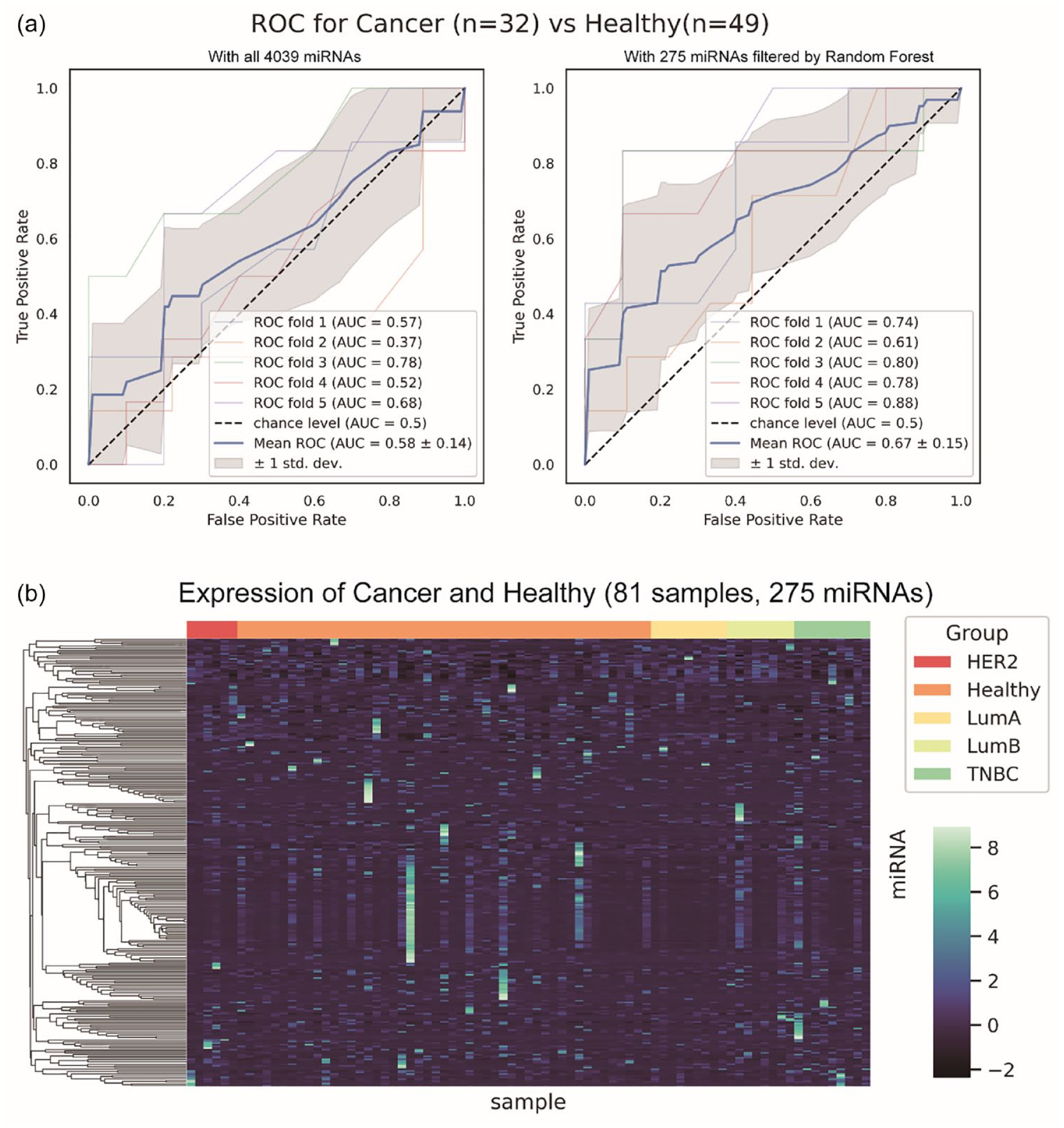
ROC curves were generated using sets of miRNAs to distinguish cancer patients from healthy individuals. (a) ROC curve analysis of all 4039 detectable miRNAs (left side) and a small subset of 275 filtered miRNAs (right side). (b) Cluster dendrogram of all samples for the 275 filtered miRNAs.
Figure 3(b) shows the heatmap showing the expression of the filtered miRNAs. The ensemble approach involving the RF algorithm produced better results than the generic statistical approach. This is probably due to the complicated or combinatorial expression patterns of miRNAs in urine. The miRNAs in urine are a mixture of different tissues and organs at their final stop, so their expression patterns are no longer obvious and are detectable by generic statistical methods.
RF algorithms with filtered miRNAs identify intrinsic subtypes of BC
Following the approach to distinguish healthy controls from women with BC, we applied RF analysis to identify miRNA patterns that would allow us to substratify patients into the distinct BC subtypes of our patient cohort: luminal A, luminal B, Her2-enriched, and TNBC.
For HER2-enriched BCs, 175 miRNAs out of the 4039 miRNAs were sufficient to increase the AUC on average from 0.55 ± 38 to 0.68 ± 0.32 (Figure 4(a), compare left to right). In the case of LumA-type cancer, differential expression of 195 miRNAs was associated with an increase in the AUC from 0.7 ± 0.32 to 0.78 ± 0.26 on average (Figure 4(b), compared left to right). As one can easily derive from the figures with ever-increasing ROCs, an increased number of runs with more samples will increase the true-positive rate dramatically.

ROC curves were generated using sets of miRNAs to distinguish subtypes of breast cancer. (a) ROC curve for HER2 patient samples versus healthy individuals using all 4039 detectable miRNAs (left side) and with only a small subset of 175 filtered miRNAs (right side). (b) ROC curve for Lum A-type breast cancer patient samples versus healthy individuals using all 4039 detectable miRNAs (left side) and with only a small subset of 195 filtered miRNAs (right side).
For luminal B-type BC, we found 191 miRNAs that distinguish Lum B-carrying patients from healthy individuals, the difference in which increased the AUC from 0.57 ± 0.2 to 0.71 ± 0.24 (Figure 5(a), left/right). Finally, TNBC was detected by RF analysis of 189 miRNAs, for which the AUC was 0.65 ± 0.31 and the AUC was 0.39 ± 0.22 for all the detectable miRNAs (Figure 5(b)). We found this to be the most dramatic increase in sensitivity among all the subgroups.

ROC curves were generated using sets of miRNAs to distinguish subtypes of breast cancer. (a) ROC curve for Lum B breast cancer patient samples versus healthy individuals using all 4039 detectable miRNAs (left side) and with only a small subset of 191 filtered miRNAs (right side). (b) ROC curve for triple-negative breast cancer patient samples versus healthy individuals using all 4039 detectable miRNAs (left side) and with only a small subset of 189 filtered miRNAs (right side).
The filtered miRNA subgroups exhibited no miRNA species overlap
Among the filtered miRNAs above, there were very few overlapping miRNAs (Figure 6(a)). There were no common miRNAs among the four subtypes. This finding suggested that the associated miRNAs might be distinct across these four subtypes. Most of these filtered miRNAs were not significantly or differentially expressed according to DGEA.

Overlapping and distinct miRNAs were identified in cancer patients and the indicated subgroups.
Discussion
BC treatment is based on tumor biology and tumor stage. Therefore, early detection has been an important step toward improving the curation rates observed over the last several decades. Today, it is common practice in industrialized countries to screen the female population for BC regularly in national programs based on mammography. Improved mammography approaches using machine learning for deeper and more accurate image analysis are therefore the next logical step to detect BC as early as possible to improve treatment and curation options.17,18 Nevertheless, the tremendous technical and timely effort, physical discomfort during the procedure, and monetary aspects of this technique could lead to the use of an easy, fast, and cost-effective prescreening method, which in the case of a positive finding would lead to an additional imaging method.
Furthermore, early information about tumor biology would likely be useful for stratifying consecutive imaging and work-up procedures.
Stratifying cancer patients based on noninvasive methods is currently a tremendous challenge. Especially in BC, the diagnosis of TNBC has much more severe implications for the patient than a luminal A-type tumor. Therefore, detecting this disease noninvasively and obtaining further information on the type of tumor would be extremely beneficial. This approach would give the treating physician a distinct advantage for subsequent work-up and treatment decisions.
Here, we present the first tightly controlled miRNA sequencing effort of urine samples from BC patients to gain insight into how the miRNA genome is regulated in this disease and its intrinsic subtypes. Earlier efforts from our group focused on specific miRNAs known to be regulated in BC using a proprietary miRNA amplification paradigm. 9 These findings already indicated that urine samples can exhibit high variability in miRNA content due to individual differences in diet, hydration status, and collection methods. Nevertheless, in the current approach, we implemented miRNA sequencing as an innovative approach for urinary analysis to understand how many miRNAs in the currently known genome are regulated in BC and whether consecutively identified signatures might represent specific subclasses of BC, allowing their detection from noninvasive urine samples.
We found the let-7-miRNA family to be strongly represented in the cancer cohort, as would be expected from studies on other cancer entities using different methods of detection. The Let7-miRNAs are dysregulated in the lung, 19 pancreatic, 20 colorectal, 21 and papillary thyroid 22 cancers and, as recently described, in BC. 23 Let7 was further shown to regulate cancer stemness. 24
Apart from these initial findings, we also detected considerable variability among the top regulated miRNAs in some samples (e.g., variability of let-7c expression in healthy individuals (Figure 2(a)), making an individual diagnosis of BC or its subclasses less reliable. We therefore applied a machine learning approach to the sequencing data to investigate whether the patterns of multiple miRNAs would be more informative than those of several strongly differentially regulated miRNAs. Interestingly, the RF approach outclassed the decision tree, logistic regression, and SVM so dramatically, making it the method of choice for future analysis of miRNA sequencing data from urine samples.
An increase or decrease in a single given miRNA did not seem to have as much impact as a whole “signature” of miRNA expression changes (Figure 1 vs Figure 3). It seems feasible that a disease like BC, affecting the whole body and eliciting multiple cellular and molecular biological changes, would lead to a wide-ranged miRNA network shift instead of regulating only a couple of individual miRNAs. The detection of very specific subsets of miRNA regulatory networks, specifically identifying both BC patients and even their specific intrinsic subtypes, is innovative and, thus far, not known. Nevertheless, more surprisingly, these patterns of miRNAs overlap very little with each other; on average, only 10%–15% of miRNAs are commonly regulated, whereas most miRNAs clearly identify a subgroup or BC in general. This, to our knowledge, has not been shown before and raises the question of whether previous data should be reanalyzed with a more unbiased approach to possibly identify yet unknown patterns. However, only a machine learning approach can unravel this issue, as has been shown in other fields of research.25 –27
While this study identifies a potential miRNA signature for BC detection, we were unable to calculate key performance indicators such as sensitivity and specificity due to the limited sample size. Future studies should include a larger, independent cohort to provide reliable estimates of sensitivity and specificity and to validate the clinical utility of the identified miRNA signature.
Consecutively, an important focus of further research should be the reduction and minimization of miRNAs included in our identified distinct miRNA pattern and an expansion of the patient cohort. The applicability of our technology for screening or early detection also relies on the sensitivity, specificity, false-positive, and false-negative rates. The optimization of these pertinent parameters relies on large cohorts of patients and healthy control samples, which will have to be analyzed for this purpose.
Conclusion
In summary, our study represents an innovative approach and “proof of principle” concept for a sensitive noninvasive, urine-based, liquid biopsy test to detect BC and its distinct intrinsic subtypes with a wide variety of application options.
Supplemental Material
sj-docx-1-tam-10.1177_17588359241299563 – Supplemental material for Random forest algorithm identifies miRNA signatures for breast cancer detection and classification from patient urine samples
Supplemental material, sj-docx-1-tam-10.1177_17588359241299563 for Random forest algorithm identifies miRNA signatures for breast cancer detection and classification from patient urine samples by Jochen Maurer, Matthias Rübner, Chao-Chung Kuo, Birgit Klein, Julia Franzen, Julia Wittenborn, Tomas Kupec, Laila Najjari, Peter Fasching and Elmar Stickeler in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
This work was supported by the Genomics Facility, a core facility of the Interdisciplinary Center for Clinical Research (IZKF) Aachen within the Faculty of Medicine at RWTH Aachen University. We thank Lothar Häberle for his advice and support.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.