
Abstract
Despite significant progress in the treatment of some types of cancer, high-grade gliomas (HGGs) remain a significant clinical problem. In the case of glioblastoma (GBM), the most common solid tumor of the central nervous system in adults, the average survival time from diagnosis is only 15–18 months, despite the use of intensive multimodal therapy. Chimeric antigen receptor (CAR)-expressing T cells, which have already been approved by the Food and Drug Administration for use in the treatment of certain hematologic malignancies, are a new, promising therapeutic option. However, the efficacy of CAR-T cells in solid tumors is lower due to the immunosuppressive tumor microenvironment (TME). Reprogramming the immunosuppressive TME toward a pro-inflammatory phenotype therefore seems particularly important because it may allow for increasing the effectiveness of CAR-T cells in the therapy of solid tumors. The following literature review aims to present the results of preclinical studies showing the possibilities of improving the efficacy of CAR-T in the TME of GBM by reprogramming the TME toward a pro-inflammatory phenotype. It may be achievable thanks to the use of CAR-T in a synergistic therapy in combination with oncolytic viruses, radiotherapy, or epigenetic inhibitors, as well as by supporting CAR-T cells crossing of the blood–brain barrier, normalizing impaired angiogenesis in the TME, improving CAR-T effector functions by cytokine signaling or by blocking/knocking out T-cell inhibitors, and modulating the microRNA expression. The use of CAR-T cells modified in this way in synergistic therapy could lead to the longer survival of patients with HGG by inducing an endogenous anti-tumor response.
Introduction
High-grade gliomas (HGGs) account for nearly 25% of central nervous system (CNS) tumors in adults. 1 Despite their rare occurrence, they pose a significant problem due to poor outcomes. 1 In the case of glioblastoma (GBM), the most common solid tumor of the CNS in adults, the average survival time from diagnosis is only 15–18 months, despite the use of intensive multimodal therapy (tumor resection, radiotherapy, and chemotherapy).2,3 It is, therefore, necessary to search for new therapeutic methods that may extend the survival rate of patients with HGGs. Chimeric antigen receptor (CAR)-expressing T cells, which have already been approved by the Food and Drug Administration for use in the treatment of certain hematologic malignancies, are a new, promising therapeutic option.3,4 However, the efficacy of CAR-T cells in solid tumors is lower due to the immunosuppressive tumor microenvironment (TME).5,6 The TME limits the effectiveness of effector T cells, thereby limiting the effectiveness of CAR-T cells, by (i) reducing the infiltration of T cells into the TME through abnormal vascular organization resulting from impaired angiogenesis and insufficient chemotactic gradient, (ii) increased local concentration of immunosuppressive cytokines, (iii) increased expression of checkpoint inhibitors on cells present in the TME, (iv) disorders of CAR-T-cell metabolism, which results from the hypoxic conditions prevailing in the TME, as well as the presence of other negative regulators of T-cell effector functions in TME (e.g., IDO1, Arg1, adenosine). 3 Considering the negative impact of the immunosuppressive TME on the functioning of CAR-expressing cells, to improve the effectiveness of therapy, strategies should be developed to reprogram the immunosuppressive TME into a pro-inflammatory TME. This can be achieved by intensifying the chemotactic gradient and normalizing impaired angiogenesis in the TME, increasing the concentration of pro-inflammatory cytokines in the TME, making CAR-T cells resistant to the action of immunosuppressive cytokines or checkpoint inhibitors, and inducing the increased activation of the innate immune system by leading to immunogenic cancer cell death via radiotherapy or the use of oncolytic viruses (OVs).7–11 This will not only improve the effector mechanisms of the CAR cells themselves but may also contribute to the influx of other cells of the patient’s immune system to the tumor site.2,5,12 The involvement of other immune cells may allow the elimination of tumor cells that do not express the CAR target antigen, thus leading to complete tumor elimination or restoration of the equilibrium phase in which the immune system controls the tumor.12,13 The following literature review aims to discuss the results of preclinical studies showing the possibilities of enhancing the efficacy of CAR-T cells in the TME of GBM by reprogramming the TME toward a pro-inflammatory phenotype. The review will discuss various strategies to overcome immunosuppressive TME based on improving CAR-T-cell entry into the TME, improving CAR-T effector functions by modifying cytokine signaling, increasing CAR-T activity by blocking or knocking out T-cell inhibitors, and modulating microRNA expression in CAR-T cells. The review will also present the benefits and limitations of synergistic therapies: combinations of CAR-T cells with OVs, radiotherapy, or epigenetic inhibitors.
Strategies to overcome immunosuppressive TME
Improving CAR-T-cell entry into the TME
Cytokine modulation
The modulation of cytokine signaling can be used to improve the entry of CAR-T cells into cancer cells as it may be impaired in the absence of an optimal chemokine gradient. 7 One of the cytokines promoting the development of GBM is interleukin (IL)-8, which increases the recruitment of tumor-associated neutrophils and myeloid-derived suppressor cells (MDSCs) to the TME, promotes angiogenesis and metastasis, and strengthens the resistance of cancer stem cells (CSC) to treatment. It should also be noted that the therapeutic methods, for example, radiotherapy, contribute to the increase in IL-8 concentration in the TME. Accordingly, Jin et al. constructed C-X-C motif chemokine receptor 1 (CXCR1)- or C-X-C motif chemokine receptor 2 (CXCR2)-modified CAR-T cells. Both CXCR1 and CXCR2 are IL-8 receptors; therefore, their co-expression by CAR-T cells will promote the recruitment of CAR-T cells to the TME. An additional advantage of this approach is the simultaneous effect of CAR-T cells on reducing IL-8 concentration. 14
Blood–brain barrier manipulation
Another approach was proposed by Huang et al. in a preclinical study. They increased the migration of CAR-T cells into the brain across the blood–brain barrier (BBB) using gastrodin (GAS), a compound derived from traditional Chinese herbs. GAS contributes to an increase in the expression of sphingosine-1-phosphate receptor 1 (S1PR1) on T cells, as well as an increase in the expression of chemokine signaling factors such as guanine nucleotide-binding protein gamma 8, C-C motif chemokine ligand 17, CXCL3, and CXCL5. This leads to an increase in the actin cytoskeleton of CAR-T cells, which supports their migration and passage through the BBB to the tumor site. 15 Increasing S1PR1 expression on T cells could also play a role in reprogramming the TME toward a more pro-inflammatory phenotype. GBM cells induce the sequestration of T cells in the bone marrow by reducing the expression of S1PR1 on the surface of T cells. Physiologically, S1PR1 binds to sphingosine-1-phosphate (S1P), the higher concentrations of which are present in the blood and lymph compared to the lymphatic organs. The resulting chemotactic gradient allows T cells to enter the circulation. The loss of S1PR1 from the surface of T lymphocytes disrupts the penetration of T lymphocytes into the circulation, dependent on the S1P-S1PR1 axis, which leads to lymphopenia in GBM patients and, as a result, the insufficient infiltration of immune cells in the tumor site. 16
In turn, Sabbagh et al. temporarily increased the permeability of the BBB to CAR-T cells using low-intensity pulsed ultrasound (LIPU). This led to the increased penetration of CAR-T cells into the CNS after 24 h (p < 0.005) and 72 h (p < 0.001) compared to mice that did not receive sonication. This resulted in increased survival durations in murine GBM models (80 vs 35 days in the control group). However, it is worth emphasizing that, on the one hand, LIPU may promote the transformation of the TME toward a pro-inflammatory phenotype by increasing the penetration of tumor antigens into the circulation and the activation of antigen-presenting cells, but on the other hand, increased BBB permeability may also increase the infiltration of the TME by immune cells with an immunosuppressive phenotype, such as regulatory T lymphocytes (Treg) or MDSCs. 17 A more selective increase in CAR-T cell permeation across the BBB was proposed by Ji et al. by modifying the CAR construct so that the extracellular domain additionally contains the rabies virus glycoprotein (RVG29). RVG29 binds to the nicotinic acetylcholine receptor present in the BBB, resulting in the significantly greater infiltration of CAR-T cells in mouse brains in the in vivo study of a mouse model of orthotopic xenograft GBM. In addition, CAR-T cells containing RVG29 had a more favorable phenotype (more central memory T cells) compared to CAR-T cells without RVG29. Nonetheless, the possibility of using RVG29 to improve the infiltration of CAR-T cells into the TME depends on further studies, which should focus on assessing the infiltration of CAR-T cells into the spinal cord and its impact on the functioning of the CNS. 18
Normalizing impaired angiogenesis
Insufficient CAR-T infiltration in the TME is caused not only by an unfavorable cytokine gradient or BBB but also by a disturbed vascular system caused by abnormal angiogenesis. Dong et al. analyzed the synergistic therapy (CAR-T and an anti-vascular endothelial growth factor (VEGF) antibodies) and showed that CAR-T-cell infiltration into the TME in a mouse model of GBM improved from 3.4% to 14.9% thanks to the use of anti-VEGF. 8 Another strategy to improve vascularization was proposed by Ma et al., who achieved the normalization of the vascular microenvironment in the TME of GBM by genetic reprogramming of tumor endothelial cells (ECs). At the first stage of the study, the authors showed that PAK4 knockout (KO) in a specially generated mouse line reduced intratumoral hypoxia and caused a change in the morphology of vessels from tortuous to organized in the TME of GBM, without affecting the physiological process of angiogenesis, proliferation, and migration of normal ECs. At the next stage of the study, the authors used the synergistic therapy consisting of a selective p21-activated kinase 4 (PAK4) inhibitor (KPT9274) and CAR-T cells and showed that the use of KPT9274 contributed to (i) reducing intratumoral hypoxia, (ii) normalizing the vascular network in the TME, (iii) reducing the expression of S100A4, ACTA2, and CDH2 genes in the TME, (iv) increasing the concentration of adhesion molecules (vascular cell adhesion molecule 1 and intracellular adhesion molecule 1 (ICAM-1)) on ECs. The normalization of vessels in the TME and an increase in the concentration of adhesion molecules improved the infiltration of CAR-T cells into the TME, while reducing hypoxia in the TME contributed to decreasing its immunosuppressive effect on CAR-T cells, thereby increasing their effectiveness—in the case of the combined therapy within 5 days after the infusion of CAR-T cells, the tumor volume in the mouse model of GBM decreased by approximately 80%. 19 Another strategy for EC normalization was proposed by Zhang et al., who indicated that the increased expression of phosphoglycerate dehydrogenase (PHGDH) in EC causes their hypertrophy, and thus promotes the formation of the hypoxic and immunosuppressive TME in GBM. The heterozygous KO of PHGDH in a mouse line specially generated for this purpose resulted in a change in vessel morphology from tortuous vessels with excessive sprouts to defined vessels with fewer sprouts in the TME. Therefore, a synergistic therapy combining the administration of PHGDH inhibitors (WQ2201) and CAR-T cells was proposed. A study on mouse models of GBM showed that the synergistic therapy caused complete tumor regression in 20% of the tested mice, and also prolonged survival in the remaining mice compared to the mice treated with monotherapy—CAR-T cells or WQ2201. In the case of the synergistic therapy, on day 34 after administering GL261 cells to mice, 30% of them were still alive, while all remaining mice (the untreated group and the group treated with monotherapy—CAR-T cells or WQ2201) died due to tumor progression. 20
Improving the distribution of CAR-T cells
To bypass the BBB, CAR-T cells are increasingly administered locoregionally or intracerebroventricularly instead of intravenously in clinical trials. 3 Ogunnaike et al. have developed a biocompatible fibrin gel encapsulating CAR-T cells which can be applied to the surgical cavity created after tumor resection. Placing a porous, biodegradable fibrin gel containing CAR-T cells in the postoperative cavity allows for the gradual biodistribution of CAR-T cells while maintaining their functionality. Fibrin gel could also contribute to modifying the TME phenotype because, in addition to the CAR-T cells themselves, it could be adapted to the gradual, local release of pro-inflammatory cytokines. 21
The aforementioned methods of improving CAR-T cell entry into the GBM TME are innovative ways of increasing CAR-T-cell infiltration in the TME of GBM. It should be noted, however, that to determine the safety profile of these methods and the possible implications of their use in clinical practice further research is mandatory.
A summary of the methods of improving CAR-T-cell entry into the GBM TME is presented in Table 1.
Methods of improving CAR-T-cell entry into the GBM TME.

BBB, blood–brain barrier; CAR-T, chimeric antigen receptor T cell; CXCR1, C-X-C motif chemokine receptor 1; CXCR2, C-X-C motif chemokine receptor 2; GBM, glioblastoma; PAK4, p21-activated kinase 4; PHGDH, phosphoglycerate dehydrogenase; TME, tumor microenvironment; VEGF, vascular endothelial growth factor.
Improving CAR-T effector functions by modifying cytokine signaling
In addition to improving CAR-T-cell entry into the TME, cytokine signaling can also be used to improve CAR-T cell effector functions. To reproduce the activation of T lymphocytes as thoroughly as possible, the fourth generation of CARs—T cells redirected for universal cytokine-mediated killing (TRUCKs) were constructed, which, apart from one co-stimulatory domain, contained an additional transgene enabling inflammatory cytokine production—see Figure 1. 9

Physiologically, T cells require three signals for activation: (1) recognition by the TCR receptor on the T cell of a specific antigen presented on the APC via the antigen-MHC complex; (2) co-stimulation via the interaction of CD80/CD86 present on the APC with CD28 present on T lymphocytes; and (3) co-stimulation via pro-inflammatory cytokines present in the local environment. The fourth-generation CAR construct most accurately produces this mechanism: (1) Signal I is induced by the interaction of the CAR present on the CAR-T cell with the CAR target antigen present on the tumor cell, (2) the co-stimulation signal is transmitted through the co-stimulatory domain, for example, CD28 present in the CAR construct, (3) an additional co-stimulatory signal via pro-inflammatory cytokines is obtained in an autocrine way thanks to the transgene present in the CAR construct, which enables the production of pro-inflammatory cytokines by the CAR-T cell.
The most frequently added transcription factors to the CAR construct allowed for the overexpression of IL-7, IL-12, IL-15, and IL-18.7,10 Increasing the concentration of pro-inflammatory cytokines in the TME may contribute to reducing the immunosuppressive effect of the TME on immune cells. For example, IL-15 supports the expansion and survival of T cells and also promotes the development of stem cell-like memory T cells, which are characterized by longer lifespan, resistance to apoptosis, and the ability to quickly differentiate into effector T cells in vivo.7,22 Krenciute et al. examined the effectiveness of the use of TRUCKs targeting the interleukin 13 receptor subunit α-2 (IL13Rα2) antigen, with an additional built-in IL-15 transcription factor (IL13Rα2-CAR.IL15-T cells) in GBM therapy. Transgenic expression of IL-15 in in vivo studies contributed to an increased effector activity, more intense proliferation, and the prolonged presence of IL13Rα2-CAR.IL15-T cells in the mouse body compared to IL13Rα2-CAR-T cells. 23 Secretory IL-15 can promote the transformation of the immunosuppressive phenotype of the TME toward a pro-inflammatory one also by binding to the IL-15 receptor α-chain present on MDSCs. Zannikou et al. constructed two types of CAR-T cells: (i) anti-IL13Rα2-CAR T cells additionally expressing IL-15 secretor (CAR.IL15s) and (ii) anti-IL13Rα2-CAR T cells in which IL-15 was fused to the CAR (CAR.IL15f), which ultimately turned out to be more effective. In in vitro study CAR.IL15f, co-cultured with MDSCs, led to a reduction in the expression of immunosuppressive IL-10, arginase-1, and transforming growth factor β (TGF-β), and in an in vivo study CAR.IL15f increased the infiltration of CD3+, CD8+ lymphocytes, natural killer (NK) cells, and B lymphocytes into the TME while reducing the infiltration of immunosuppressive cells originating from the bone marrow. 24 Another cytokine considered is IL-7, which promotes T-cell proliferation and the formation of memory T cells. Due to the limited half-life of IL-7, as well as its insufficient concentration in the TME, Swan et al. modified CAR-T cells to enable them to secrete IL-7 as well as FMS-like tyrosine kinase 3 ligand (Flt3L). Flt3L is a key factor in the development and expansion of dendritic cells (DCs), therefore increasing its concentration in the TME may contribute to increasing the efficiency of antigen presentation by DCs, thereby increasing the specific antitumor response of CD8+ T cells. In vivo studies in a mouse model of GBM showed that the use of CAR-T cells secreting IL-7 and Flt3L increases the infiltration of CAR-T cells themselves, as well as DCs and CD8+ T cells in the TME of GBM, and thus may promote the pro-inflammatory phenotype of TME. 25 Another potent pro-inflammatory cytokine is IL-12. Meister et al. generated mouse CAR-T cells expressing mouse IL-12 and mouse interferon α2 (IFNα2). In a study in immunocompetent orthotopic glioma-bearing mice, CAR-T cells co-expressing IL-12 and IFNα2 contributed to reprogramming the TME toward a more pro-inflammatory phenotype by increasing the infiltration of bystander CD4+ and CD8+ T cells. In addition, CAR-T cells co-expressing IL-12 and IFNα2 were characterized by highly expressed genes associated with T-cell activation, and cells expressing only CAR were characterized mainly by the expression of genes associated with exhaustion. 26 However, it has been shown that modifying T lymphocytes to enable them to secrete IL-12 may result in life-threatening systemic toxicity in some patients. 27 Therefore, Agliardi et al. assessed the effectiveness of the local, intratumoral administration of a single dose of IL-12 with CAR-T cells. A study in a mouse model of GBM showed that the synergistic therapy significantly improved mice survival, and the CAR-T-cell population in the mice treated with IL-12 showed lower expression of lymphocyte-activation gene 3 and programmed cell death protein 1 (PD-1), compared to the CAR-T-cell population in the mice not treated with IL-12. 28 The study also showed that IL-12 contributes to the reprogramming of the TME to be more pro-inflammatory by (i) increasing the infiltration of CD4+ T cells in the TME, (ii) reducing the infiltration of Treg lymphocytes in the TME, (iii) increasing the expression of major histocompatibility complex (MHC) class II molecules in microglia, (iv) increasing programmed death-ligand 1 (PDL-1) expression on the tumor-associated monocyte population, and (v) decreasing arginase 1 concentration in the TME. A study in a mouse model also showed that the intratumoral administration of IL-12 did not cause serious systemic effects in mice (transient, moderate increase in interferon γ (IFN-γ), no increase in IL-6 and granulocyte-macrophage colony-stimulating factor). 28 Another alternative to obtaining a supportive signal from immunostimulatory cytokines without simultaneously increasing their systemic concentration in the body is to construct a T cell that simultaneously expresses CAR and constitutively signals cytokine receptors on its surface. Shum et al. constructed T cells co-expressing CAR targeting the disialoganglioside (GD2) and constitutively active IL-7 receptor variant (IL7R), activating the signal transducer, and activator of transcription 5 pathway (GD2-CAR.C7R-T cells). An in vivo study showed that GD2-CAR.C7R-T cells were more effective than GD2-CAR T cells against neuroblastoma cells in a mouse xenograft model. A similar strategy was tested in a mouse model of GBM. The control group received 104 ephrin type-A receptor 2 (EphA2)-CAR T cells, while the study group received 104 EphA2-CAR.C7R T cells. CAR-T cells without C7R co-expression were unable to eliminate the tumor at such a low dose, unlike EphA2-CAR.C7R T cells, indicating a C7R-dependent improvement in the persistence and effector functions of CAR-T cells. 29 Another method to obtain a supportive signal from immunostimulatory cytokines was proposed by Woodell et al., who generated therapeutic second-generation induced neural stem cells platform—human-induced neurospheres (hiNeuroS) secreting C-C motif chemokine ligand 5 (CCL5)/IL-15 (hiNeuroSRANTES-IL-15). The use of the synergistic therapy involving the administration of hiNeuroSRANTES-IL-15 and CAR-T cells in a murine model of orthotropic GBM xenografts led to durable tumor remission for 60 days post-treatment in all animals studied, while the MS of mice treated with CAR-T cells alone was 19 days. The therapeutic effect of the synergistic therapy was related to enhanced T-cell migration and persistence achieved by the secretion of CCL5 and IL-15 into the TME by hiNeuroS. 30
In addition to increasing the concentration of pro-inflammatory cytokines in the TME, further improvement of the effectiveness of CAR-T cells can be achieved by reducing the concentration of immunosuppressive cytokines. Li et al. generated TGF-β-trapped anti-epidermal growth factor receptor variant III (EGFRvIII) CAR-T cells that contain the TGF-β type II receptor (TGFRII) ectodomain, making them resistant to the action of TGF-β. The use of CAR-T cells modified in this way led to the increased secretion of C-C motif chemokine ligand 4 and tumor necrosis factor α (TNF-α) in the TME, as well as to the polarization of microglia toward the pro-inflammatory M1 phenotype. 31 Another action taken to limit the impact of immunosuppressive cytokines present in the TME on the effector functions of CAR-T cells is the construction of chimeric cytokine receptors, which, despite binding to the anti-inflammatory cytokine, activate signal plates characteristic of pro-inflammatory cytokines. The co-expression of CAR and chimeric cytokine receptors on the surface of T lymphocytes increase their cytolytic activity and proliferative capacity. 10
The role of cytokines in combination immunotherapy is largely limited by dose-dependent systemic adverse events. Due to the short half-life of cytokines in serum, high, repeated doses of cytokines are required to achieve a therapeutic effect. This may ultimately lead to a life-threatening systemic inflammatory response, the clinical picture of which may include hypotension, fever, acute renal failure, respiratory failure, and neuropsychiatric symptoms.32,33 This limitation in the use of cytokines in clinical practice can be overcome by local delivery of cytokines to the tumor, thereby achieving antitumor effects while limiting systemic side effects. The previously described approaches to a large extent allow for the local delivery of cytokines. It should be noted, however, that leakage during the production of the inducible cytokines by the CAR-T cells may lead to a systemic toxic reaction, and therefore, before implementing the aforementioned solutions in clinical trials, attention should be paid to the safety profile of the therapy. 32 If side effects occur, the activation of inducible caspase 9, previously transduced into CAR-T cells, would lead to the apoptotic death of CAR-T cells, thus improving the safety profile of the therapy. 10
A summary of the methods of improving CAR-T-cell effector functions by modifying cytokine signaling is presented in Table 2.
Methods of improving CAR-T-cell effector functions by modifying cytokine signaling.
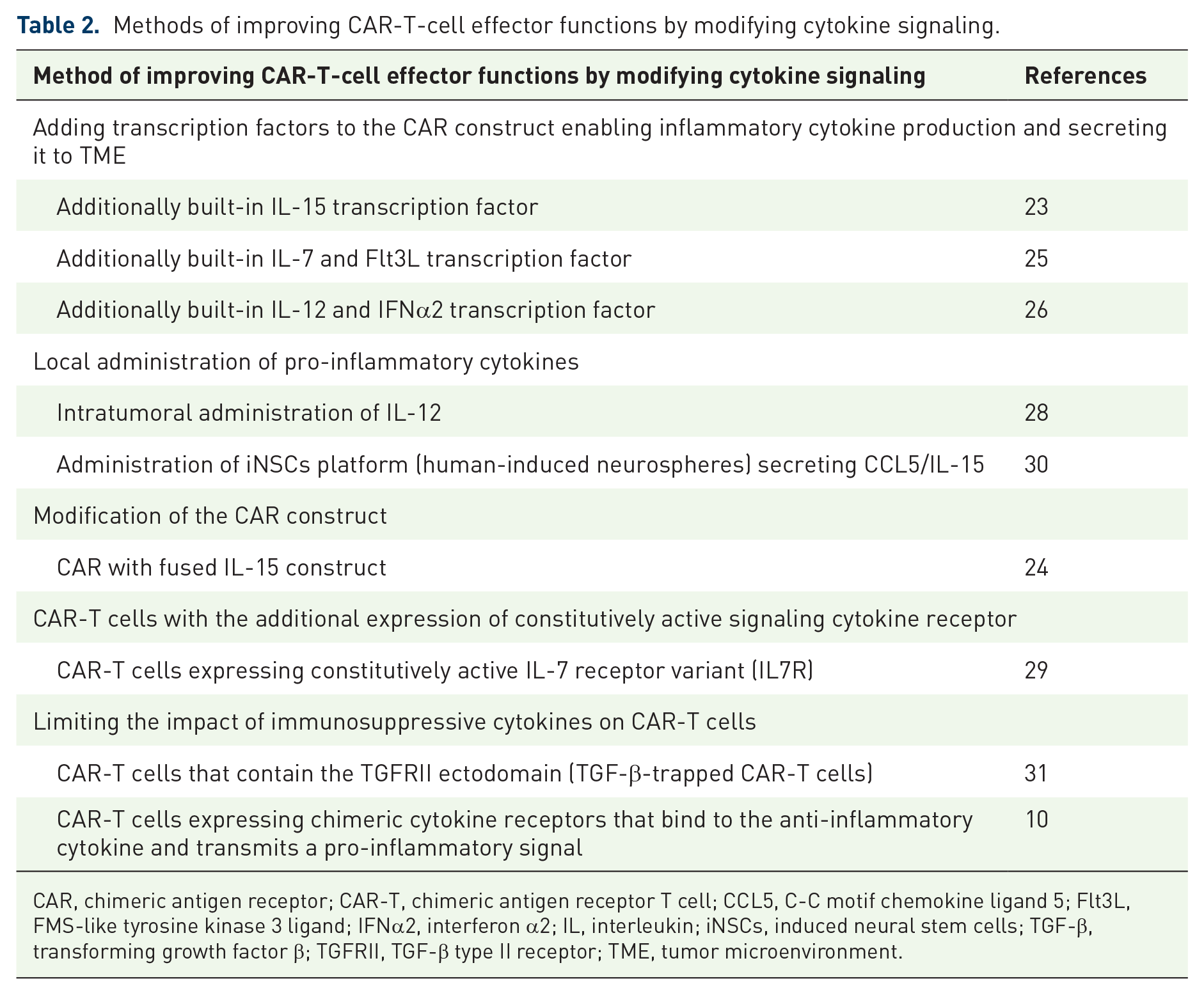
CAR, chimeric antigen receptor; CAR-T, chimeric antigen receptor T cell; CCL5, C-C motif chemokine ligand 5; Flt3L, FMS-like tyrosine kinase 3 ligand; IFNα2, interferon α2; IL, interleukin; iNSCs, induced neural stem cells; TGF-β, transforming growth factor β; TGFRII, TGF-β type II receptor; TME, tumor microenvironment.
Improving CAR-T activity by blocking or knocking out T-cell inhibitors
Under physiological conditions, checkpoint inhibitors prevent the excessive activation of the immune system and autoimmune diseases. However, the increased expression of checkpoint inhibitors in the TME contributes to the impaired anti-tumor immune response.34,35 It has been shown that PD-1 is expressed by CAR-T cells, and the degree of PD-1 expression on cells present in the infusion preparation is directly proportional to progression-free survival. 11 Therefore, reducing the expression of checkpoints, blocking them, or knocking them out could improve the effector functions of CAR-T cells.
One of the most commonly used approach to blocking the PD-1/PDL-1 axis is the use of the synergistic therapy combining the administration of CAR-T cells with anti-PD-1 antibodies. Zhang et al. investigated the effect of administering different doses of CAR-T cells with or without nivolumab (dose: 10 mg/kg) on the survival of mice with orthotropic GBM transplantation. All mice that received 20 × 105 CAR-T cells, regardless of the use of nivolumab, died before day 30 of the experiment, probably due to toxicity induced by a high dose of CAR-T cells. In turn, all mice treated with CAR-T cells at doses of 10 × 105 and 5 × 105 in monotherapy died by day 40 of the experiment. However, 50% of mice treated with the synergistic therapy with nivolumab and CAR-T cells at doses of 10 × 105 or 5 × 105 were still alive on day 50 of the experiment. The optimal therapeutic effect was achieved using the synergistic therapy (10 × 105 CAR-T cells with nivolumab), which allowed the survival of mice to be extended to day 60 of the experiment. 36 A similar study was conducted by Song et al., who blocked PD-1 present on EGFRvIII CAR-T cells using anti-PD-1 antibodies. Similarly to the aforementioned studies, CAR-T cells with PD-1 blockade were characterized by longer in vivo survival compared to CAR-T cells without blockade. In addition, it was observed that in the GBM xenograft mice treated with CAR-T cells with PD-1 blockade, the infiltration of CD3+ T cells in the tumor tissue significantly increased, which may indicate a pro-inflammatory transformation of the TME. 37 An increase in the effectiveness of CAR-T cells in combination with anti-PD-1 antibody was also observed by Shen et al., who in their study showed that synergistic therapy increases the secretion of IL-2 and IFN-γ by CAR-T cells in vitro, and also significantly raises the cytotoxicity of CAR-T cells compared to the use of CAR-T cells in monotherapy. 38
Another approach to the problem of the increased expression of checkpoint inhibitors in the TME is the possibility of converting their inhibitory signal into a co-stimulatory signal thanks to immune checkpoint modulation. One approach is to design armored CAR-T cells that co-express CAR and a PD-1/cluster of differentiation (CD28) fusion protein. After PDL-1 binds to PD-1, thanks to the intracellular signaling domain of CD28, the T lymphocyte receives a co-stimulatory signal instead of an inhibitory signal. 39
In turn, Zhu et al. constructed EGFRvIII-targeted CAR-T cells with PD-1 KO and showed that upon activation they secreted more IFN-γ and IL-2 when compared to CAR-T cells without PD-1 KO. In an in vivo study on a mouse model of GBM, CAR-T cells with PD-1 KO showed greater effector functions on GBM cells and were also characterized by longer in vivo survival compared to CAR-T cells without PD-1 KO. 40 Similar results indicating a beneficial effect of PD-1 KO on effector functions were obtained by Nakazawa et al., who constructed a third-generation CAR-T with tumor necrosis factor receptor superfamily 9 (4-1BB) and CD28 co-stimulatory domains, directed against the EGFRvIII antigen with PD-1 KO. 41 Choi et al., in addition to PD-1 KO, also deleted the genes encoding endogenous T-cell receptor and beta-2 microglobulin. Thanks to this modification, it was possible to obtain an allogenic CAR T-cell product, which may be important in the case of patients in whom, due to lymphopenia, it was not possible to isolate a sufficient number of T lymphocytes to create a good quality autologous CAR-T-cell preparation. 42 A summary of the mechanisms by which the impact of the PD-1/PDL-1 axis on CAR-T cells can be limited is presented in Figure 2.

The PD-1/PDL-1 axis, by inactivating the activating signal transmitted by CAR upon antigen binding, inhibits the effector functions of CAR-T cells. To override the unfavorable effect of the PD-1/PDL-1 axis on CAR-T cells, one can (a) induce low expression or knock-out of PD-1 from CAR-T cells. (b) Use anti-PD-1 antibodies, which, by binding PD-1, will block the possibility of PD-1 interacting with its ligand. (c) Generate a fusion protein (e.g., PD-1/CD28), which, after binding the ligand, transmits a co-stimulatory signal instead of an inhibitory signal thanks to the activation domain.
Another approach was presented by Sengupta et al. who, by inhibiting glycogen synthase kinase 3 (GSK-3) in T cells, reduced the PD-1 expression on CAR-T cells. GSK-3 regulates the expression of transcription factor T-box (T-BET) in activated T cells. Since the increased expression of PD-1 in T cells is dependent on the decreased expression of T-BET, the pharmacological inhibition of GSK-3 using a GSK-3 inhibitor (SB216763) increases CAR-T cell resistance to exhaustion. In addition, it should be noted that, physiologically, GSK-3 is constitutively active in naïve T cells. During T-cell activation, GSK-3 is transiently inactivated by phosphorylation, which is associated with the rapid, clonal proliferation of T cells. GSK-3 phosphorylation is, however, temporary, and its reactivation results in the clonal contraction of T cells. Therefore, the use of SB216763 also resulted in the promotion of the effector memory phenotypes of CAR-T cells and reduced the activation-induced cell death of CAR-T cells. Experience has also shown that the synergistic therapy with CAR-T cells and SB216763 can support long-term tumor control by CAR-T cells. After the second administration of tumor cells to mice that had previously been treated with the synergistic therapy, no new tumors developed in any of the four animals, while in the group treated only with CAR-T cells, after the repeated injection of tumor cells, new tumor foci developed in all animals. 43
However, the PD-1/PDL-1 axis is not the only inhibitor of CAR-T cells effector functions. Yin et al. checked the validity of using cytotoxic T-cell antigen 4 (CTLA-4), T-cell immunoglobulin and mucin domain 3 (TIM-3), and PD-1 blockade as a possibility to improve the effector functions of CAR-T cells directed against IL-13Rα2 and EGFRvIII. At the initial stage of the study, the expression of checkpoints was assessed on anti-IL-13Rα2 CAR-T cells and anti-EGFRvIII CAR-T cells co-cultured with tumor cells. Specifically, increased CTLA-4 expression occurred primarily on anti-IL-13Rα2 CAR-T cells, whereas increased PD-1 and TIM-3 expression was more pronounced on anti-EGFRvIII CAR-T cells. The aforementioned result was correlated with the therapeutic effect of the synergistic therapy combining the administration of CAR-T cells and checkpoint-targeted antibodies. Anti-EGFRvIII CAR-T cells combined with anti-CTLA-4 showed no increase in activity, while CTLA-4 blockade had the greatest benefit with anti-IL-13Rα2 CAR-T cells. In turn, the administration of anti-PD-1 and TIM-3 antibodies resulted in the greatest increase in anti-EGFRvIII CAR-T-cell activity. Depending on their specificity, CAR-T cells therefore required different blockades to improve their effectiveness in the same tumor model, which was predictable based on the result of checkpoint expression on CAR-T cells co-cultured with tumor cells, indicating the need for personalized therapy also in terms of selecting appropriate checkpoint inhibitors for CAR-T cells. 44
Improving the effector functions of CAR-T cells by modulating microRNA expression
Micro ribonucleic acid (miRNA) is a single-stranded, non-coding molecule composed of 19–23 nucleotides. It is estimated that the human genome encodes information from about 900 to over 1900 different miRNAs, which, in turn, influence the expression of over 30% of human protein-coding genes. Each miRNA is complementary to a specific fragment of messenger RNA (mRNA), and fusing with this fragment regulates its translation.45,46 The inhibition of mRNA translation is carried out by the miRNA-inducing silencing complex (miRISC), which, in addition to miRNA, consists of the Argonaute protein (AGO). MiRNA present in miRISC binds to complementary mRNA sequences (miRNA responsive elements (MREs)). Depending on the degree of complementarity of miRNA binding to MREs, mRNA can be (i) cleaved by the AGO2 endonuclease or (ii) mRNA translation can be inhibited by the miRISC complex (translational repression), which is achieved by accelerating the degradation of the polyA chain at the 3′ end of mRNA and mRNA decapping. Deadenylation and decapping of mRNA ultimately enable mRNA degradation by exoribonucleases present in cells.47,48
One of the miRNAs that could improve the functioning of CAR-T cells in the TME is miR-17–92, which promotes a cellular response with a Th1 phenotype. 49 CD4+ T cells with a Th1 phenotype exhibit antitumor activity by secreting IL-2, TNF-α, and IFN-γ, while CD4+ T cells with a Th2 phenotype promote tumor progression.50,51 In patients with GBM, CD4+ cells are polarized in the TME toward an unfavorable Th2 phenotype with reduced miR-17-92 expression. This process is additionally intensified by the lymphocytes with the Th2 phenotype themselves by secreting IL-4, which reduces the expression of miR-17-92 in T lymphocytes, leading to a decrease in the Th1/Th2 ratio, which is an unfavorable prognostic factor.49–51 Therefore, increased miR-17-92 expression in CAR-T cells could support their effector functions in the TME. However, in T lymphocytes collected from GBM patients to introduce the CAR construct into them, a decreased expression of miR-17-92 was demonstrated. In addition, glucocorticosteroids administered during the GBM therapy also reduced the level of miR-17-92 expression in T lymphocytes. 52 Therefore, Ohno et al. introduced an additional transgene containing miR-17-92 into CAR-T cells to increase its expression in these cells. To introduce the transgene into CAR-T cells, Ohno et al. constructed a lentiviral vector for miR-17-92 (FG12-EF1a-miR-17/92). The vector contained two promoters: EF1a, which controlled the expression of miR-17-92, and UbiC, which controlled the expression of green fluorescent protein (EGFP), allowing the assessment of transduction efficiency. This was observed both when cells were exposed to increasing concentrations of temozolomide (a chemotherapy drug used in the treatment of GBM, which has an inhibitory effect on T-cell proliferation) and without it. In the next step, in an in vivo study on immunocompromised mice, the long-term stability of CAR-T cells co-transduced with miR-17-92 was assessed. On day 7 after i.c. inoculation of GBM cells, 10 mice received CAR-T cells (3C mice) with temozolomide and another 10 mice received CAR-T cells additionally co-transduced with miR-17-92 (3C+miR mice) with temozolomide. No statistically significant difference was observed in the survival of mice treated with CAR-T cells (nine mice from the first group and eight mice from the second group survived longer than 40 days). On day 49 after CAR-T cell infusion, four 3C mice and three 3C+miR mice were re-injected, i.c. with GBM cells to the opposite hemisphere. Re-administration of tumor cells resulted in tumor growth in all four 3C mice, but no tumor growth occurred in 3C+miR mice. Increasing the effectiveness of long-term tumor control by CAR-T cells additionally co-transduced with miR-17-92 may result from increasing the survival of CAR-T cells and increasing the secretion of IFN-γ by CAR-T cells. 52
Synergistic therapy: CAR-T with OVs
OVs are one of the therapeutic methods under consideration that could significantly improve the functioning of CAR-T cells in the TME. 53 OVs, by multiplying and then causing the oncolysis of tumor cells, both positive and negative for the expression of the CAR target antigen, can to some extent prevent tumor escape associated with the low or zero expression of the CAR target antigen. This is an example of targeted therapy because OVs can replicate in and lyse only cancer cells without damaging normal cells. OVs also enhance the response of the innate immune system because damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) are emitted in connection with the oncolysis of cancer cells.9,54 In addition, OV-infected cancer cells reduce the expression of MHC class I molecules on their surface, which contributes to the activation of NK cells. 55 OVs also contribute to the formation of tumor-associated antigens, which, when presented in a complex with MHC, can stimulate a specific immune system response. The intensification of inflammation caused by infecting cancer cells with OVs contributes to a reduction in the infiltration of immunosuppressive cells (e.g., Tregs and MDSCs) and an increase in the concentration of pro-inflammatory cytokines in the TME (e.g., type I IFN and CXCL10—C-X-C motif chemokine ligand 10).55,56 Type I IFN may promote activation, proliferation, differentiation, and survival of CD8+ T cells in the TME. In addition, it promotes favorable polarization of CD4+ lymphocytes toward the Th1 phenotype and increases the expression of co-stimulatory molecules (CD40, CD86) on the surface of lymphocytes.55–57 In turn, CXCL10 is a chemotactic factor for T cells, so an increase in its concentration in the TME may lead to the increased infiltration of the TME by CAR-T cells.55–57 Reducing the infiltration of the TME by immunosuppressive cells will translate into a reduction in the concentration of immunosuppressive cytokines (e.g., IL-10 and TGF-β), which have an inhibitory effect on the effector functions of CAR-T cells. 55 OVs pleiotropically support the reprogramming of the immunosuppressive TME toward a pro-inflammatory phenotype and thus may increase the effectiveness of CAR-T cells.
In a preclinical study, Zhu et al. investigated the combination therapy of anti-cluster of differentiation 70 (CD70) CAR-T cells and oncolytic herpes simplex virus-1 (oHSV-1). Infecting GBM cells with oHSV-1 significantly increased the levels of the pro-inflammatory cytokines and at the same time decreased the levels of the anti-inflammatory cytokines. This led to the reprogramming of the TME to a more pro-inflammatory phenotype, and thus to the increased infiltration of immune cells (including CAR-T cells). The dual therapy was significantly more effective in killing cancer cells than each therapy alone. 58
In addition, OVs can be genetically modified by inserting transgenes into them so that when they multiply in cancer cells, they will cause the release of molecules that enhance the effector functions of CAR-T cells into the immunosuppressive TME. The aforementioned findings indicate the possibility of reversing the immunosuppressive TME by OV—see Figure 3. 9

OVs can modulate the immunosuppressive TME toward a pro-inflammatory phenotype by (1) inducing immunogenic cell death and thereby leading to (2) activation of the innate immune system. (3) In addition, thanks to the inserted transgenes, cancer cells infected by OVs can secrete immunomodulators that contribute to the increased infiltration of the TME by immune cells and the improvement of their effector functions.
Wang et al. examined cancer cells infected with the specially constructed C-X-C motif chemokine ligand 11 (CXCL11)-armed oncolytic adenoviruses (oAds-CXCL11) and proved that infected cancer cells released CXCL11 into the TME. CXCL11 is a ligand of the C-X-C motif chemokine receptor 3 (CXCR3) receptor found on T cells and it plays a role in creating a chemotactic gradient allowing for the effective infiltration of T cells into the site of inflammation. After transduction of the CAR construct into T cells, CAR-T cells have been shown to additionally increase CXCR3 on their surface. Therefore, the synergistic therapy of oAds-CXCL11 with CAR-T cells should increase CAR-T-cell infiltration in the TME of GBM. A study in a mouse model of GBM showed that four out of five mice treated with the synergistic therapy achieved a durable antitumor response and survived until the end of the study (over 70 days). Meanwhile, in the case of CAR-T therapy alone, two out of five mice survived to day 35 of the experiment, and then died due to tumor progression before day 40 of the experiment. In addition, it was shown that the use of oAds-CXCL11 contributed to the reprogramming of the immunosuppressive TME into an immunostimulatory TME by (i) increasing the infiltration of CD8+, CD3+, CD45+ T lymphocytes in the TME, (ii) increasing the infiltration of NK cells in the TME, (iii) increasing the infiltration of macrophages with a pro-inflammatory M1 phenotype in the TME, (iv) reducing MDSCs infiltration in the TME, and (v) reducing Treg lymphocyte infiltration in the TME. 54 Due to the beneficial effect of IL-7 on the effector functions of CAR-T cells, Huang et al. constructed an interleukin-7-loaded oncolytic adenovirus (oAD-IL7). Four of five GBM xenograft mice treated with the synergistic therapy (oAD-IL7 and anti-B7 homolog 3 protein (B7H3) CAR-T) survived to the end of follow-up (day 60), whereas the mice treated with only oAD-IL7 survived fewer than 40 days after tumor cell administration and the mice treated with only anti-B7H3 CAR-T fewer than 50 days after tumor cell administration. 59
To increase the effectiveness of CAR-T cells, Wing et al. inserted another transgene into the OV allowing for a reduction in the possibility of tumor escape from CAR-T-cell surveillance caused by the loss of the CAR target antigen. They constructed an oncolytic adenovirus armed with a bispecific T-cell engager (OAd-BiTE) targeting epidermal growth factor receptor (EGFR). BiTE secreted from virus-infected cancer cells redirects CAR-T cells and T lymphocytes to EGFR+ cancer cells. Thanks to this, CAR-T cells can effectively eliminate cancer cells negative for the CAR target antigen, as long as they express EGFR. 60
The advantage of OVs therapy is its safety profile. One of the frequently reported side effects occurring in patients after the administration of OVs is mild fever, and toxicity after the administration of OVs is most often described as grade 1–2. 61 However, further studies are required to determine the safety profile of the synergistic therapy combining the administration of OVs and CAR-T cells. The activation of the immune system caused by the administration of OVs may intensify the side effects of CAR-T therapy, which include cytokine release syndrome, neurotoxicity, off-target toxicity, and anaphylaxis. 62 To ensure a better safety profile of the therapy, in the case of the simultaneous administration of CAR-T cells with other preparations that stimulate the immune system, it would be beneficial to use inducible safety switches allowing the induction of apoptosis of CAR-T cells in the event of severe side effects of the therapy. 10
Synergistic therapy: CAR-T with radiotherapy
One of the synergistic therapies being considered is also the use of CAR-T cells with radiotherapy. Radiotherapy of the tumor site may increase the expression of MHC class I molecules on tumor cells and thus may contribute to a more effective presentation of antigens to immune system cells. In addition, DAMPs are released from cells destroyed by ionizing radiation (IR), which may contribute to the greater activation of innate response cells and, thanks to the pro-inflammatory cytokines released by them, facilitate the infiltration of the TME by CAR-T cells. 9 Wang et al. constructed CAR-T cells targeting B7-H3 and examined their effectiveness in combination with the use of radiotherapy. Pre-irradiation increased the expression of ICAM-1, B7-H3, and fas cell surface death receptors on tumor cells, which promotes the infiltration of the TME by CAR-T cells and also sensitizes tumor cells to the antitumor activity of CAR-T cells. In addition, in an in vivo study of a mouse model of glioma, it was observed that the mice treated with the combination therapy had a greater infiltration of T lymphocytes in the TME, which may indicate the pro-inflammatory reprogramming of the TME due to the release of DAMPs and PAMPs from cancer cells as a result of radiotherapy. 63 Further confirmation of the efficacy of the synergistic CAR-T-cell therapy with radiotherapy was presented in the paper by Murty et al. Using a mouse model of GBM and intravital fluorescence microscopy, they showed that radiotherapy enabled faster extravasation of CAR-T cells from the vessels and their local expansion in the tumor stroma. The use of CAR-T cells without whole-body irradiation in mice resulted in the insufficient infiltration of CAR-T cells within the tumor stroma, thus preventing complete tumor regression. It should be emphasized that a complete antitumor response was achieved both in the case of whole-body irradiation of mice and with focal radiotherapy limited to the tumor site. The study results indicate that radiotherapy may not only increase the effector functions of CAR-T cells but it may also support their migration inside the TME. 64
On the other hand, it has been shown that IR may contribute to the formation of the immunosuppressive TME by activating the signal transducer and activator of transcription 1 (STAT1)-interferon regulatory factor 1 (IRF1)-ectonucleoside triphosphate diphosphohydrolase-1 (CD39) axis in glioma CSCs. The IR-induced increased expression of CD39 on cancer cells leads to CD39-dependent degradation of one of the DAMPs—extracellular adenosine triphosphate (eATP). The use of IR together with anti-CD39 antibodies prevents the level of eATP from being reduced, making it possible to reprogram the immunosuppressive TME into a pro-inflammatory TME—see Figure 4. Under normal conditions, eATP binds to the purinergic P2X7 receptor present in DCs, which makes them produce more pro-inflammatory cytokines, IL-1β and IL-18. By combining IR with anti-CD39, the process of immunogenic cell death is promoted, which can additionally stimulate CAR-T cells in vivo via the T-cell receptor (TCR). 65 The mechanism of action of the combined therapy is presented in Figure 4.
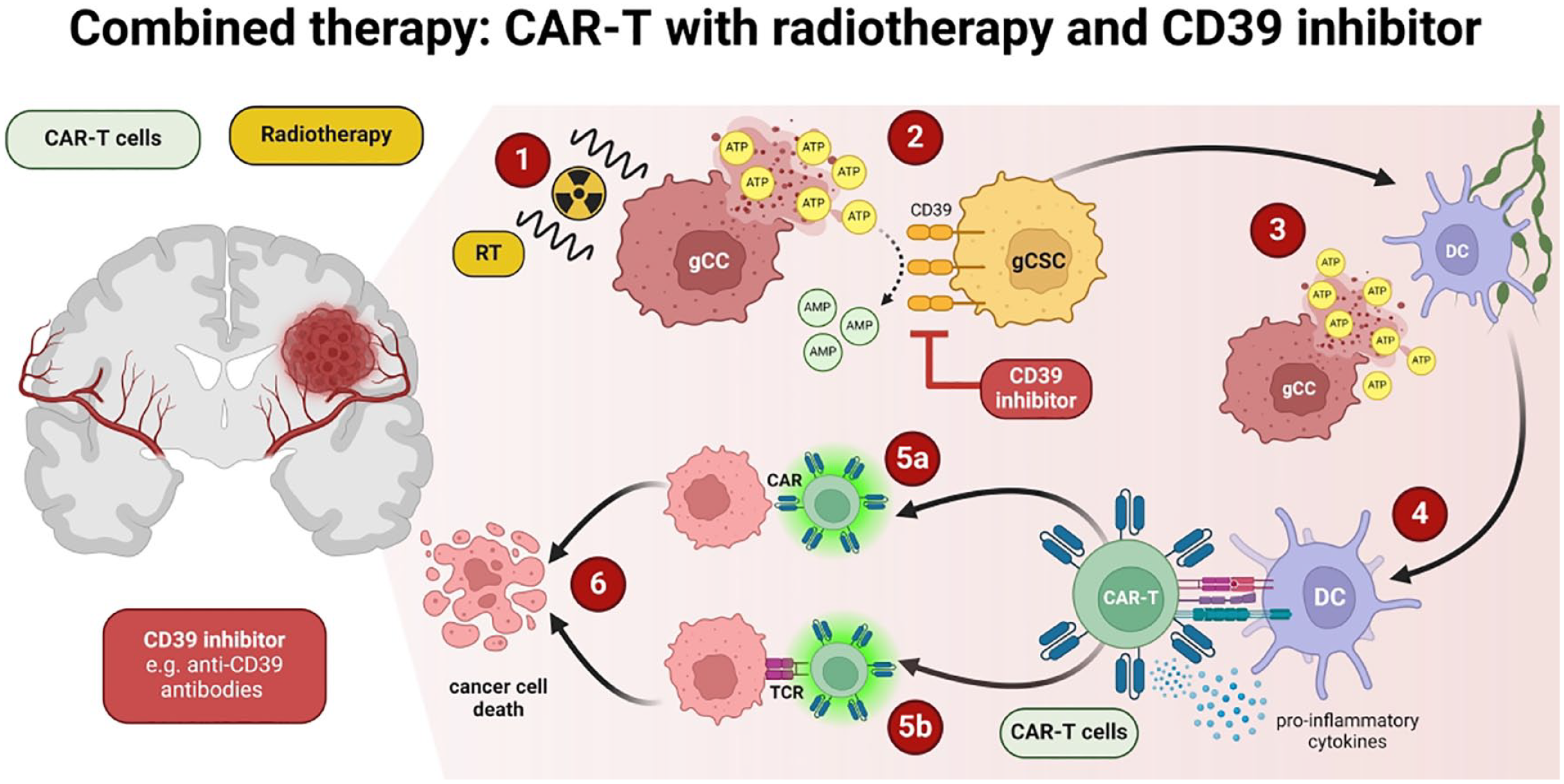
Mechanism of action of the combined therapy (CAR-T cells, radiotherapy, CD39 inhibitors): (1) The use of radiotherapy causes the release of ATP (one of DAMPs) from the glioma cancer cells, (2) thanks to the use of CD39 inhibitor, ATP is not broken down into AMP by CD39 present at glioma cancer stem cells, (3) ATP released from glioma cancer cells activates DCs, (4) DCs activate the TCR receptor present on CAR-T cells, thanks to which CAR-T cells by (5a) binding CAR to the target antigen and (5b) by binding TCR with the antigen present on cancer cells lead to (6) cancer cell death.
Synergistic therapy: CAR-T with epigenetic inhibitors
In response to the presence and activity of CAR-T cells in the TME, GBM cancer cells activate the transcription of genes encoding immunosuppressive molecules, thereby increasing (i) the expression of T-cell inhibitors on their surface, (ii) the local concentration of cytokines that may have an immunosuppressive effect in the TME (IL-6, IL-8, IL-1β), and (iii) indoleamine 2,3-dioxygenase 1 concentration, thus contributing to the impaired functioning and exhaustion of CAR-T cells in the TME, resulting in the development of resistance to CAR-T cell treatment. Xia et al. in their study partially reversed this process by inhibiting bromodomain containing 4 (BRD4), which is one of the regulators of the transcription of immunosuppressive genes in cancer cells at the epigenetic level. The use of a BRD4 inhibitor (JQ1) blocks the binding of BRD4 to methylated lysine, thereby interfering with the activation of gene expression and intensifying the immunosuppressive phenotype of the TME (PD-L1, PD-L2, HVEM, GAL9, IL6, IL8, CSF2, BIRC3, IDO1, and IL1B). The use of the synergistic therapy in a mouse model of GBM—JQ1 and CAR-T cells resulted in a 43% reduction in the expression of genes encoding immunosuppressive molecules, which led to more effective inhibition of tumor growth by CAR-T cells and prolonged mice survival.66,67 The development of resistance after a transient response to the use of JQ1 in GBM monotherapy has been reported, similar to the case of the use of CAR-T cells alone. 66 Preclinical results of the synergistic GBM therapy combining CAR-T and JQ1 administration are promising; however, further research is necessary to determine the potential implications and challenges of applying such therapies in clinical practice.
Other approaches to improve CAR-T-cell functions
One of the physiological inhibitors of T-cell function is diacylglycerol kinase (DGK). DGK exists in several isoforms and catalyzes the conversion of diacylglycerol (DAG) to phosphatidic acid. In physiological conditions, TCR activation triggers a signaling cascade, and one of its elements is DAG which mediates the activation of the activating protein-1(AP1)/nuclear factor of the activated T-cell (NFAT) pathway, which ultimately leads to the activation of the T lymphocyte. In the case of DAG deficiency, the NFAT pathway is activated, which causes the lymphocyte to enter a state of anergy. 68 Therefore DGK, by participating in the inactivation of DAG, may contribute to the weakening of the effector functions of CAR-T cells, especially since the overexpression of the alpha isoform of DGK (DGKα) has been demonstrated in CAR-T cells. 69 DGKα is involved in regulating the functioning of T lymphocytes by influencing cell proliferation and migration as well as regulating their activation. In turn, the ζ isoform of DGK (DGK ζ) is involved in regulating signaling with TCR, toll-like receptor, and high-affinity IgE receptor. 68 Accordingly, Jung et al. constructed three types of anti-EGFRvIII CAR-T cells (139 CAR-T) with the KO of DGKα (αKO), DGKζ (ζKO), or DGK double KO of DGKα and DGKζ (dKO). It was shown that 139 dKO CAR-T cells had better effector functions than single KO CAR-T cells because they (i) secreted more pro-inflammatory cytokines (IFN-γ and IL-2), (ii) had enhanced TCR signaling (extracellular signal-regulated kinase-1 phosphorylation was more intense and lasted longer in them), (iii) proliferated after repeated stimulation with tumor antigens (they did not quickly enter the state of anergy), (iv) retained their effector functions in the presence of immunosuppressive TGF-β and prostaglandin E2, and (v) increased the infiltration of lymphocytes into the TME. In addition, DGK KO promoted the transformation of CAR-T cells into a more favorable effector memory population phenotype. 70 It was also observed that the treatment of 139 CAR-T cells with αKO, ζKO, or dKO only minimally improved the effector functions of 139 CAR-T cells with αKO, leading to a slight increase in IFN-γ secretion; and an additional KO of PD-1 with 139 CAR-Ts had no effect on their function. 70 Another example of an inhibitory effect on T-cell effector functions is protein phosphatase 2A (PP2A). This enzyme works by inhibiting the mTORC1 signaling pathway. Therefore, blocking the action of PP2A may lead to the increased activity of the mTORC1 pathway, and thus it may contribute to increasing the effectiveness of T cells. Cui et al. checked whether the use of the PP2A enzyme inhibitor—LB-100 can increase the effectiveness of CAR-T cells. In their study, the authors constructed CAR-T cells directed against carbonic anhydrase IX (CAIX) and tested them in vitro and in vivo on various GBM cell lines, both as a monotherapy and in combination with LB-100. They found that inhibiting PP2A enzyme function significantly increased both CAR-T-cell cytotoxicity and survival in mouse models of GBM. 71 Increasing the effectiveness of CAR-T cells in the immunosuppressive TME can also be achieved using lenalidomide (LENA) promoting the ubiquitination of the transcription factors Ikaros (encoded by IKZF-1) and Aiolos (encoded by IKZF-3). Ikaros and Aiolos, by binding to the promoter region of the IL-2 gene, silence gene expression. Therefore, the degradation of Ikaros and Aiolos contributes to the increased secretion of IL-2 from T cells after TCR stimulation. During the in vitro study, CAR-T cells combined with LENA secreted more IL-2, TNF-α, IFN-γ, and their proliferation also increased. An additional advantage of LENA is its anti-angiogenic effect, which could normalize impaired angiogenesis in the TME, as well as its ability to inhibit immunosuppressive Treg cells and enhance the effector functions of NK cells.72,73 Since the systemic administration of LENA may be associated with side effects, Zou et al. generated CAR-T cells with KO of the IKZF3 which led to the upregulation of genes related to the activation, proliferation, and cytotoxic functions of T cells. However, it is worth noting that genes related to T-cell apoptosis were also upregulated, which may be related to shortening the lifespan of CAR-T cells. 73
The metabolism of cancer cells also influences the shaping of the TME. They respirate mainly anaerobically, breaking down glucose into lactate, and this process is controlled by lactate dehydrogenase (LDH). The formed lactates, through epigenetic modification (histone lactylation), regulate gene transcription both in cancer cells and immune cells. In this way, high lactate concentrations may modify the effector functions of CAR-T cells infiltrating the TME. In a study by Sun et al., the use of oxamate—an inhibitor of the LDHA subunit of LDH in combination with CAR-T cells—was investigated. It has been shown that high lactate concentration increases the lactylation of histone H3K18, which increases the activity of CD39, CD73, and CCR8 gene promoters. Limiting this phenomenon by the presence of oxamate leads to changes in the TME, mainly the reduction of the infiltration of immunosuppressive Treg lymphocytes, and to changes in the CAR-T cells themselves, including increasing the expression of IFN-γ, perforin, and granzyme B, which enhances their cytotoxic activity. 74 In turn, Durgin et al. indicate that on the surface of cancer cells of solid tumors are sialic acid molecules that exhibit immune checkpoint activity. The authors proposed the use of Clostridium perfringens neuraminidase to remove sialic acid residues from tumor target cells, which could translate into better effector functions of CAR-T cells. It turned out that the use of CAR-T cells secreting neuraminidase not only affects immune checkpoints but also causes the cells to differentiate into naïve T lymphocytes, which leads to a better control of tumor growth in in vivo studies. 75
The most numerous cell populations in HGG-TME (30%–50% of cells) contributing to tumor progression are tumor-associated macrophages.76,77 Macrophages in the TME may present a pro-inflammatory M1 phenotype or an immunosuppressive M2 phenotype, and depending on factors present in the environment, the same cell may display an M1 or M2 phenotype. 78 Glioma cells constitutively secrete factors that promote the polarization of the macrophage phenotype toward M2, and therefore macrophages produce only small amounts of pro-inflammatory cytokines.76,77 Macrophages with the M2 phenotype limit the effectiveness of the anticancer effect through secreted chemical compounds. 76 In a study by Yang et al., the authors used a small molecule compound—toosendanin (TSN)—to change the phenotype of macrophages to a pro-inflammatory one. The study results indicate that TSN inhibits Hck and Lyn kinases responsible for the immunosuppressive nature of macrophages in the TME. Therefore, the use of TSN reprograms the TME to a more pro-inflammatory phenotype, favoring the use of targeted immunotherapies such as CAR-T cells. 79
Conclusion
Therapeutic approaches that target the shift of the immunosuppressive TME toward a pro-inflammatory phenotype contribute to improving the effectiveness of CAR-T cells in vitro and in vivo. However, further research on the TME itself should be conducted because a full understanding of the relationships between the cells that make up the TME is necessary for the appropriate selection of personalized therapies, which may ultimately lead to the improved survival of patients with HGGs.