
Abstract
Over the last 15 years, immunotherapy has revolutionised treatment paradigms and improved outcomes in a range of malignancies. Despite these advances, the role of immunotherapy in standard prostate cancer (PCa) management is limited, and Sipuleucel-T is the only approved immunotherapeutic agent. This article reviews the role of checkpoint inhibitors (ICIs), T-cell engagers (TCEs) and chimeric antigen receptor (CAR)-T cells in PCa treatment. Phase II/III trials of ICIs as monotherapy or combination treatment have been negative to date. Early phase data for TCE are promising, but the feasibility of adoption of TCEs into the clinic will depend on overcoming neutralising anti-drug antibodies and limiting toxicities. CAR-T cells have demonstrated feasibility and acceptable safety profiles in early phase clinical trials, and it is hoped that the ongoing development of later-generation constructs and therapeutic combinations will enhance outcomes.
Plain language summary
Over the past 15 years, traditional forms of immunotherapy have revolutionised survival from some ‘hot’ cancers, such as melanoma and other skin cancers. Immune cell ‘cold’ cancers, such as prostate cancer, are resistant to traditional immunotherapy. Here, we explain some of the reasons behind the failure of traditional immunotherapy in prostate cancer. We also review new types of immunotherapy which look promising as ways to overcome resistance and harness the immune system against prostate cancer cells. We suggest that newer forms of immunotherapy are worthy of ongoing development in the treatment of prostate cancer.
Keywords
Introduction
Globally, prostate cancer (PCa) is the second most common cancer in men and the fifth leading cause of cancer mortality, accounting for an estimated 397,000 deaths annually. 1 Whilst there have been significant advances in treatment, there remains a need for new therapies to reduce this burden of disease.
Recent advances in management of metastatic PCa
In the past 15 years, changes in treatment have significantly improved survival and quality of life for men with metastatic PCa. Multiple therapeutic classes have driven these improvements in clinical outcomes including androgen receptor pathway inhibitors (ARPIs), poly ADP-ribose polymerase inhibitors (PARPi), taxanes and radiopharmaceuticals. The ARPIs (abiraterone, apalutamide, darolutamide and enzalutamide) have been approved for various indications across the advanced PCa spectrum.2–13 Approved radiopharmaceuticals include Lutetium-177 [177Lu]-prostate-specific membrane antigen (PSMA)-617 (177Lu-PSMA-617) 14 and radium 223 15 for metastatic castration-resistant prostate cancer (mCRPC). Monotherapy with PARPi such as olaparib 16 and rucaparib, 17 and combinations involving ARPIs and PARPi18,19 have shown efficacy and achieved regulatory approval for mCRPC with patients with homologous recombination deficiency (HRD), and recently updated results from the TALAPRO-2 trial met the key secondary endpoint of overall survival (OS) in unselected patients. 20 Taxane chemotherapy remains relevant, with docetaxel used either in upfront triplet therapy for metastatic hormone-sensitive prostate cancer (mHSPC) or in the subsequent treatment of mCRPC and cabazitaxel in mCRPC following progression on docetaxel. 21
Despite the progress in treatment, patients with metastatic PCa ultimately develop resistance to these treatments and are most likely to die from their cancer.
The role of immunotherapy in current PCa management
Over the same period, other solid tumours have had an explosion of success with immunotherapies, in particular immune checkpoint inhibitors (ICIs). Early success in melanoma 22 and non-small cell lung cancer 23 has been followed by the adoption of ICIs as standard first-line management for a large range of advanced solid malignancies. 24 There are four broad categories of immunotherapy agents: (i) ICIs, (ii) T-cell-engaging therapies, (iii) genetically modified T cells, in particular, chimeric antigen receptor-T (CAR-T) cells and (iv) cancer vaccines. ICIs act by blocking negative regulatory proteins and thus allowing host T-cell activation. They rely on the immune infiltrate present within the cancer to facilitate the immune response. 25 In contrast, T-cell-engaging therapies and CAR-T cells act to redirect cytotoxic T cells to predefined tumour targets, to facilitate primarily major histocompatibility complex (MHC)-independent cancer cell elimination. 26
The efficacy of immunotherapy varies widely between tumour types, in part due to the tumour immune micro-environment (TIME). The TIME consists of tumour-derived components, including tumour cells and chemokines; and host-derived components, including immune cells, humoral factors, stroma and paracrine feedback loops. 27 The PCa TIME exhibits a ‘cold’ immunological phenotype. The key tumour-derived alterations in PCa affecting the TIME include loss of PTEN protein expression, decreased expression of MHC class I and low tumour mutational burden (TMB). 28 The stroma is typically dense and sparsely populated by immune cells. 29 The sparse cellular infiltrate is skewed towards an immunosuppressive phenotype rich in regulatory FOXP3+ T cells, M2 macrophages and myeloid-derived suppressor cells. 30 In contrast, cytotoxic CD8+ lymphocytes are infrequent, dysfunctional and exhausted. 31 These cellular changes become more exaggerated throughout the natural history of PCa toward castration resistance. 32
Other relevant host factors affecting the TIME include therapeutic androgen blockade and the epidemiological effects of advanced age. The net effect of these tumour- and host-derived factors is an immune-desert, ‘cold’ TIME phenotype. 33
Sipuleucel-T
The only approved immunotherapy in PCa is the therapeutic vaccine Sipuleucel-T. Sipuleucel-T is an autologous cellular immunotherapy. It involves a collection of the autologous white blood cell-fraction containing antigen-presenting cells, exposure of the cells to prostatic acid phosphatase to induce an immune response, and reinfusion of the cells to induce a cytotoxic response against PCa cells (Figure 1). 34 The IMPACT trial of men with mCRPC with bone or nodal masses and a chemotherapy-free interval of ⩾3 months showed improved OS for those treated with Sipuleucel-T compared to placebo (median OS 25.8 months vs 21.7 months, hazard ratio (HR) 0.78, 95% confidence interval (CI) 0.61–0.98, p = 0.03). 35 Side effects of Sipuleucel-T include mild to moderate chills, pyrexia and headaches. This trial included a highly selected population; only 20% had received prior chemotherapy, 75% had a Gleason score ⩽7, most were asymptomatic and 43% had ⩽5 sites of bone metastases. There is no evidence of benefit in patients with visceral metastases or aggressive variant prostate cancer. The National Comprehensive Cancer Network guidelines recommend Sipuleucel-T be considered as initial therapy for asymptomatic or minimally symptomatic patients with mCRPC, or in men with mCRPC who have had prior treatment with docetaxel or an ARPI, but not both. 36 Usual markers of benefit (decline in prostate-specific antigen (PSA) or improvement in bone or CT scans) are not seen. Whilst it is approved by the U.S. Food and Drug Administration (FDA), it has not been approved in other jurisdictions, including Australia, the United Kingdom and Europe, and is therefore unavailable in these areas.

Categories of immunotherapeutic strategies. (a) TCEs are MHC-independent constructs directly linking a range of target TAAs with an immune effector. Current TCEs are ‘off-the-shelf’ constructs typically linking TAAs with CD-3+ T cells. (b) ICIs facilitate MHC-dependent T-cell activation by blocking immunosuppressive signalling pathways. ICIs are antigen-agnostic, nonspecific immunotherapies dependent on the net immunogenicity of the TIME rather than a specific TAA. (c) CAR-T cell therapy is also MHC-independent. Current CAR-T cell constructs are manufactured by ex vivo activation and expansion of patient-derived effector T cells, which have been genetically modified to recognise a target TAA of choice. (d) Sipuleucel-T is an autologous APC vaccine produced by ex vivo exposure of APCs to prostatic acid phosphatase, facilitating MHC-dependent T-cell activation.
Checkpoint inhibitors
ICIs have transformed the treatment of many cancers, including melanoma, lung and renal cancers, and can induce deep and enduring responses. Most current ICIs inhibit cytotoxic T lymphocyte-associated protein-4 (CTLA-4), programmed cell death protein-1 (PD-1) and programmed cell death ligand-1 (PD-L1), which hinder signalling pathways that suppress the immune response (Figure 1). Resistance to ICIs can occur due to intrinsic factors rendering primary resistance (i.e. poor response at initiation of therapy) or through dynamic changes in the TIME resulting in emergent resistance during therapy. 37 High PD-L1 expression and TMB are predictive biomarkers of ICI response in routine clinical use. 33 These changes are rare in PCa, and in addition to the ‘cold’ TIME, may be one explanation for the low response rates (RRs) seen with ICI therapy. Additional factors noted in a review by Claps et al. are the low immunogenicity of androgen deprivation therapy (ADT)-induced apoptosis, high rates of chronic corticosteroid exposure and patient selection in clinical trials. 38 The only current approved indication for ICI therapy in PCa is the tumour agnostic listing for mismatch repair deficient (dMMR) or microsatellite instability-high (MSI-H) tumours, which comprise approximately 3% of all PCa. 39 Trials for ICI therapy alone and in combination for mCRPC are outlined below.
CTLA-4 inhibition monotherapy in mCRPC
ICI activity in PCa was first investigated with the CTLA-4 inhibitor, ipilimumab.40–42 A dose escalation study of ipilimumab with or without radiotherapy showed PSA decline and radiological disease control across multiple doses and objective responses at high doses of 10 mg/kg. 43 The phase III CA184-043 trial went on to examine ipilimumab following bone-directed radiotherapy in patients with mCRPC who had progressed on docetaxel chemotherapy, 44 and found no difference in the median OS compared with placebo on primary analysis. Similarly, a study of ipilimumab versus placebo in patients who were chemotherapy naïve, minimally symptomatic, and without visceral disease, demonstrated a statistically significant longer progression-free survival (PFS) in the ipilimumab arm (5.6 vs 3.8 months), higher PSA response (23% vs 8%) and longer time to cytotoxic therapy, but no difference in OS. 45 Whilst this data seemed to suggest no effect on survival with single-agent CTLA-4 inhibition, further long-term follow-up of CA184-043 demonstrated a piecewise HR of 0.66 (95% CI 0.52–0.84) beyond 12 months, and reported cases of long-term remission.46,47
PD-1/PD-L1 inhibitor monotherapy in mCRPC
PD-L1 expression on primary PCa tissue is associated with an increased risk of biochemical recurrence. 48 Whilst PD-L1 expression on untreated PCa cells is low, the frequency of PD-L1 positivity is much higher in mCRPC and tends to be heterogeneous within a patient. 49 Tumour-agnostic trials with nivolumab monotherapy did not show activity in the PCa cohort. 50 Early phase trials of atezolizumab and pembrolizumab, however, were slightly more promising, with some PSA responses seen in the mCRPC cohorts, and objective response rates (ORRs) of 4% and 17.4% respectively.51,52
Following these signals, the phase II KEYNOTE-199 trial assessed pembrolizumab monotherapy in 3 parallel cohorts of patients with mCRPC, who had previously been treated with chemotherapy and an ARPI; PD-L1 positive (cohort 1), PD-L1 negative (cohort 2) and bone predominant PD-L1 unselected disease (cohort 3). 53 Only 6% of patients had a confirmed PSA response, and the ORR was only 5% and 3% in cohorts 1 and 2 respectively. The treatment also had toxicities; 15% of patients had grade ⩾3 treatment-related adverse events (TRAEs), including two treatment-related deaths. On exploratory biomarker analysis, a TMB and PD-L1 combined proportion score was associated with improved PSA response and longer time to PSA progression, however, numbers were too small to evaluate the effect on ORR, disease control rate or OS. 54
Combination drug therapies with ICIs
Combining ICIs with other therapeutics has been trialled across various tumour types to enhance overall tumour response. This is now the standard of care in advanced non-small cell lung cancer. 55 Given the poor RRs to single-agent ICI, combination treatment has been extensively explored based on the hypothesis that cytotoxic chemotherapy or ARPIs could enhance the antigenicity of the PCa cells and turn the ‘cold’ TIME ‘hot’. A list of key studies of combination treatments with ICIs is shown in Table 1.
Trials evaluating immune checkpoint inhibitors combined with systemic therapies.

PSA50 RR is the proportion of patients achieving ⩾50% reduction in PSA from baseline, confirmed on subsequent samples. ORR and PFS were measured using RECIST v1.1 criteria, or alternatively the Prostate Cancer Clinical Trials Working Group 3 modified RECIST v1.1. Chemotherapy naïve: no prior chemotherapy for mCRPC.
AE, adverse event; ARPI, androgen receptor pathway inhibitor; C + A, cabozantinib plus atezolizumab; CI, confidence interval; HR, hazard ratio; HRR, homologous recombination repair; mCRPC, metastatic castration-resistant prostate cancer; mo, months; ORR, objective response rate; OS, overall survival; PD-L1, programmed cell death ligand-1; PFS, progression-free survival; PSA, prostate-specific antigen; rPFS, radiographic progression-free survival; RR, response rate; TRAE, treatment-related adverse event.
Dual checkpoint blockade
Combination CTLA-4 and PD-1 inhibition have become standard-of-care in several tumour types, including melanoma, renal cell cancer and malignant pleural mesothelioma. 24 The relative doses of each drug used in combination differs by tumour type, with an acknowledgement of increased toxicity from the combination approach. As monotherapy, ipilimumab increases CD4 and CD8 T-cell infiltration into prostate tumours, and also increases expression of PD-L1 on tumour and immune cells. 71 This suggests that upregulation of other inhibitory mechanisms may be limiting disease response. A combination of CTLA-4 and PD-1/PD-L1 inhibitors attempts to overcome this. Both ipilimumab/nivolumab and durvalumab/tremelimumab have been evaluated in mCRPC (Table 1). ORRs ranged from 16% to 25%56,57 and it is suggested that efficacy is higher in chemotherapy naïve patients, those with higher TMB, HRD positivity and PD-L1 positivity. 57 Expansion of the post-chemotherapy cohort of the CheckMate-650 trial, with additional randomisation to cabazitaxel or ipilimumab monotherapy arms, demonstrated no convincing improvement of PFS or OS in dual checkpoint blockade. 58
Combination ICI plus chemotherapy
Taxanes are the standard chemotherapy in mCRPC, with docetaxel demonstrating a PSA RR of 45%, and significant improvement in OS in the pre-ARPI era. 72 Treatment with taxanes may improve immune responses to malignancy, by enhancing antigen presentation, decreasing numbers of Treg cells whilst increasing cytotoxic T cells, promoting secretion of inflammatory cytokines and stimulating monocytes to differentiate to M1 macrophages, 73 thus resulting in a synergistic effect when combined with immunotherapy. Phase I/II studies showed PSA RRs of 34%–47%, ORR of 23%–40% and median OS of 18.2–20.2 months, but phase III studies have failed to demonstrate improved PFS or OS with chemoimmunotherapy compared to chemotherapy alone (Table 1).
Combination ICI plus ARPI
The combination of ICI plus ARPI has also been explored. Androgen Receptor expression positively correlates with PD-L1 expression, 48 and progression on an ARPI may enhance immune activity by increasing levels of circulating PD-L1/2+ dendritic cells, PD-1+ T cells and NK cells, whilst reducing myeloid-derived suppressor cells.74,75 Phase I/II trials of combination ARPI and ICI have shown PSA RRs of 19%–24%, ORR 11%–25% and median OS of 20–22 months, with similar toxicity rates to early phase trials of combination chemotherapy and ICI (Table 1).
Three phase III trials, totalling over 3000 participants, were conducted to further evaluate this combination. The IMbassador250 63 and KEYNOTE-641 64 trials evaluated patients with mCRPC. Both trials included substantial numbers of patients who had previously received an ARPI, with participants switching to the alternative ARPI (ARPI-switch) with or without an ICI. Both trials were stopped due to futility, despite inactive control arms. Responses were enriched in patients with PD-L1 ⩾5%, higher CD8+ T-cell infiltration, higher TMB (⩾2.52 mut/mb) and in patients with expression of genes related to pre-existing immunity (i.e. antigen presentation, T-cell activation and recruitment, interferon signalling), but these were small subgroups. 63
In contrast, the KEYNOTE-991 trial 65 was conducted in patients with mHSPC on continuous ADT, and excluded previous ARPI use, with the intervention arm involving ICI plus ARPI versus ARPI alone. This was also ceased early due to futility (Table 1).
Overall, these data suggest a lack of additive benefit of ICI to ARPI by either its own mechanism of action or by a synergistic effect in either the castration-sensitive or castration-resistant setting.
Combination ICI plus PARPi
Alterations in homologous recombination repair pathways occur in approximately 20% of PCa, 76 including approximately 12% with germline mutations. 77 PARPi are currently approved by the FDA for use in patients with HRD mCRPC or mutations in BRCA genes, following progression on an ARPI. Treatment of cancer cells with PARPi up-regulates PD-L1 expression in vitro 78 and activates stimulator of interferon genes (STING) signalling in both BRCA-mutated and BRCA-wildtype tumours, 79 suggesting a rationale for combining with ICIs. Trials studying this combination again have been disappointing, with modest PSA and radiographic objective responses seen in unselected populations, and no improvement in combination PARPi plus immunotherapy over ARPI-switch.66–68 Subgroup analysis of the phase III KEYLYNK-010 trial, however, did suggest a potential radiographic PFS and OS benefit in patients with BRCA, but not other HRD (Table 1) mutations. 68 This highlights that biomarker selection is key, and it remains unclear whether the benefit seen in these subgroups was simply due to the PARPi use (which is now standard care in BRCA-mutated PCa) or if there was an added benefit of immunotherapy.
Combination ICI plus tyrosine kinase inhibitors
With disappointing results for ICI combined with standard therapies for mCRPC, combination with therapies not typically used in PCa have also been considered. Tyrosine kinase inhibitors (TKIs) are efficacious alone and in combination with ICIs in other cancers, such as renal cell carcinoma. 80 The TAM kinase family, targeted by the TKI cabozantinib, is overexpressed in PCa and is thought to contribute to an immunosuppressive TIME. 81 A phase Ib trial of a combination of cabozantinib with atezolizumab demonstrated a PSA RR and ORR of 23%. 82 TRAEs were similar to other combination strategies. The phase III CONTACT-2 trial further investigated this combination versus ARPI-switch, and whilst there was a statistically significant 2-month PFS benefit favouring the ICI and cabozantinib combination, there was no difference in OS (Table 1). A survival benefit was seen in some subgroups, in particular, those with liver metastases. 70
Overall, these data suggest that the combination of ICI with any systemic agent has, to date, failed to improve outcomes in mCRPC.
Multimodality therapies with ICIs
Radiation to sites of disease is another approach to enhancing the immune response. The phenomenon (known as the ‘abscopal effect’) of regression of non-target sites of disease after radiotherapy has been attributed to the triggering of a systemic immune response. 83 The previously mentioned CA184-043 trial, which demonstrated potential long-term survival benefits after giving ipilimumab following a single dose of bone-directed radiotherapy to patients with mCRPC,44,46 supports this phenomenon, however, has not been replicated or adopted into routine practice.
Radiopharmaceuticals allow for targeted delivery of radiation to widespread disease. Radium-223 is a calcium-mimetic radioactive isotope directed towards bone metastases, that induces DNA damage and improves OS in mCRPC. 84 A phase Ib study combining atezolizumab with Radium-223 showed a low ORR of 6.8% and median radiographic PFS and PSA progression of 3 months each. 85 Furthermore, 45% of patients experienced a serious adverse event, and there were four treatment-related deaths. A phase II study comparing Radium-223 with pembrolizumab to Radium-223 alone failed to demonstrate improved immune cell infiltration on repeat biopsies in the combination group, and there was no improvement in clinical outcomes. 86
More recently, 177Lu-PSMA-617 has demonstrated improved survival outcomes in patients with mCRPC who have PSMA-positive disease on gallium-68-labelled PSMA-11 positron emission tomography. 14 Two phase I studies have investigated the safety of combining ICIs with 177Lu-PSMA-617. The PRINCE trial combined pembrolizumab with up to 6 cycles of 177Lu-PSMA-617 therapy, followed by maintenance pembrolizumab for up to 2 years, and showed promising clinical activity, with a PSA RR of 76%, median radiologic PFS of 11.2 months and median OS of 17.8 months, with no new safety concerns. 87 Aggarwal et al. demonstrated that a single dose of 177Lu-PSMA-617, followed by maintenance pembrolizumab, had a favourable safety profile, a PSA RR of 56%, and a median radiological PFS of 6.9 months. 88 Randomised studies with comparator arms are required to determine the contributory efficacy of immunotherapy to 177Lu-PSMA-617.
Predictive biomarkers for ICI therapy
Despite the overall lack of efficacy demonstrated in the above trials, cases of long-lasting responses to ICIs have been reported in mCRPC. There are no reliable biomarkers to select these patients and guide ICI use. PD-L1 expression on tumour cells was established early in the use of ICIs to correlate with treatment efficacy 89 of PD-1/PD-L1 targeting agents, though measurement technique, cut-off levels and utility vary across trials and tumour types.
Early phase studies in PCa showed PD-L1 expression was associated with increased ORR, PSA RR and OS,53,57 however, this did not translate to an OS benefit in the phase III IMbassador250 trial of atezolizumab plus enzalutamide. 63 Furthermore, PD-L1 expression is low in mCRPC compared with other tumour types, and only ~30% of patients are PD-L1 positive. 49
TMB-high confers increased sensitivity to ICIs in other solid tumours by increased neoantigen burden, which can be targeted by the immune system. TMB in PCa is low, with a median TMB of 2.6 muts/Mb, and only 5% of cases have high TMB.76 There is no consistent association between TMB-status and PCa outcomes with ICI treatment.54,57,59
MSI-H or dMMR phenotypes occur in 3%–4% of PCa.39,76 In retrospective reviews, PSA50 responses in patients with dMMR disease were shown to occur in a far greater proportion of patients (65%) than in unselected PCa cohorts. 90
Biallelic cyclin-dependent kinase 12 (CDK12) loss of function mutations (6% of PCa)76,91 also result in increased neoantigen load as well as increased T-cell infiltration, and are thought to enhance immunogenicity. These genomic changes are associated with PSA responses of 11%–40%, but the absolute number of patients studied is small.92–94
The single-arm phase II INSPIRE trial was a biomarker-selected trial in mCRPC, including 69 participants with PCa that was dMMR, TMB-high (defined as ⩾7.1 muts/Mb), BRCA2-mutated or biallelic CDK12-inactivated. 95 Patients were administered a combination of nivolumab and ipilimumab therapy, followed by nivolumab maintenance therapy. Overall, 40% achieved disease control at 6 months, 47% had a PSA50 response and 38% an OR, with 25% of patients having their PSA decline to <0.1 ng/mL. The most impressive results were seen in the dMMR subgroup, with a PSA50 RR of 86%, ORR of 75% and median PFS of 32.7 months (median OS not reached). TMB-high, BRCA-mutated and CDK12-inactivated cohorts had PSA50 RRs of 38%, 30% and 20% respectively, and median PFS of 3.8, 3.5 and 1.6 months. This demonstrates strong evidence for the use of ICI in dMMR tumours, whereas TMB-high, BRCA mutations and CDK12 inactivations are not supported as predictive biomarkers.
Finally, recent translational data presented at the 2025 ASCO Genitourinary Cancers Symposium explored the role of ctDNA assays. Fizazi et al. examined the baseline and cycle 6 tumour fraction (TF) of patients enrolled in the phase III CheckMate 7DX trial of docetaxel plus nivolumab or placebo in mCRPC. In both treatment arms, high baseline TF (TF ⩾ 4.6%), and failure of TF clearance (C6D1 TF > 1%), negatively correlated with OS outcomes. The prognostic association of baseline TF outperformed prior chemotherapy or prior ARPI exposure. Whilst not yet established in routine practice, these data indicate the potential role of ctDNA as a reliable prognostic biomarker which may be considered as a stratification factor in future trials of immunotherapy in mCRPC, and as an early indicator of treatment response for chemotherapy and chemoimmunotherapy in mCRPC. 96
T-cell engaging therapies
T-cell engaging therapies include Bispecific- and Trispecific T-Cell Engagers (BiTEs and TriTES), Immune Mobilising T-cell receptors Against Cancer (ImmTACs), CAR-T and CAR-NK cells. To date, BiTEs and CAR-T cells are the most developed TCE therapies in PCa and are the focus of this review.
Bispecific T-cell engagers
T-cell recognition of antigens is usually dependent upon antigen presentation via the MHC. The interaction between T-cell receptors and MHC molecules triggers complex downstream signalling pathways, resulting in T-cell activation. T-cell activation is dependent on the affinity of the T-cell receptor for the MHC complex. 97
BiTEs are double-ended antibodies linking an effector and a target. The most commonly employed effector is a single-chain variable region antibody to the ubiquitous T-cell marker cluster of differentiation 3 (CD3), allowing for the recruitment of a T cell-mediated immune response. This is linked to the target (a single-chain variable region antibody with affinity for a tumour-associated antigen (TAA) of choice) by a protein fragment. The aim of this linkage is to induce T-cell-mediated cytotoxic tumour cell death. 98
BiTEs allow for T-cell activation independent of the MHC molecule. Crucially, the affinity of BiTEs for TAAs is higher than conventional MHC-dependent antigen recognition, such as that seen in normal physiology, and traditional immunotherapeutic classes, including checkpoint inhibitors and monoclonal IgG antibodies. As early as 2005, it was shown in cellular models that this resulted in significantly more efficient tumour cell lysis, particularly in models with low ratios of T cells. 99 Thus, BiTEs represent a promising immunotherapeutic class for ‘cold’ immune-desert TIMEs such as PCa. In addition, the MHC-independent mechanism allows for ‘off the shelf’ production (Figure 2).

BiTEs and prostate cancer TAAs.
PCa tumour-associated antigens
There are several TAAs strongly associated with PCa. These include PSMA, prostate stem cell antigen (PSCA), human kallikrein 2 (henceforth referenced in this review as hK2, sometimes known as KLK2), six transmembrane epithelial antigens of the prostate 1 (STEAP1) and transmembrane protein with EGF-like and two follistatin-like domains 2 (TMEFF2). 100 Whilst these individual antigens do not provoke a sufficient immunological response to sway the TIME from ‘cold’ to ‘hot’, they represent potential therapeutic targets for BiTEs because they are specifically expressed in tumour cells. 100
PSMA-targeting BiTEs
The first BiTEs investigated in PCa utilised PSMA as a target. Friedrich et al. reported promising preclinical results for the first-in-class anti-PSMA/anti-CD3 BiTE in 2012, 101 and the drug, which would become known as Pasotuxizumab, progressed through the pipeline.
The Pasotuxizumab experience illustrates the promise and the limitations of BiTEs in advanced PCa. A phase I dose escalation study treated patients with either daily subcutaneous (SC) injection at 11 dose levels between 0.5 and 172 μg daily, or continuous intravenous infusion at 5 dose levels between 5 and 80 μg daily. 102 This trial enrolled 68 men with previously treated mCRPC refractory to ⩾1 taxane and abiraterone or enzalutamide.
Immunogenicity and the formation of neutralising anti-drug antibodies (ADAs) limited drug exposure to SC Pasotuxizumab, highlighting what is a common pharmacokinetic issue within the class and necessitating early termination of the SC arm of the study. Root cause analysis identified problems with both the drug formation (relating to non-tolerant T-cell epitopes within the Pasotuxizumab amino acid sequence) and the route of administration. 103 Pharmacokinetics were challenging owing to a short half-life, which necessitated cumbersome daily SC injection or continuous infusion. 102
The efficacy of intravenous Pasotuxizumab was dose-dependent. Patients achieved a median best PSA response of −5% in the 5 μg daily cohort and −55% in the 80 μg daily cohort. Eighty-three percent of patients in the 40 and 80 μg cohorts experienced G3+ treatment-emergent adverse events (TEAEs), with the most common G3+ TEAEs being cytopaenias, fatigue, cytokine-release syndrome, fever and electrolyte disturbances. The toxicity profile is consistent with other agents in the class. 102
Whilst Pasotuxizumab has not entered clinical practice, subsequent medications in this class remain under investigation. A summary of PSMA-targeting BiTEs is presented in Table 2. Whilst RRs in these heavily pre-treated patients with mCRPC are promising, the high rate of cytokine-release syndrome has hampered development.
PSMA-CD3 BiTE monotherapy trials.

PSA50 RR: the proportion of patients achieving ⩾50% reduction in PSA from baseline, confirmed on the subsequent sample.
ADAs, anti-drug antibodies; ARPI, androgen receptor pathway inhibitor; BiTE, bispecific T-cell engager; CD3, cluster of differentiation 3; CRPC, castration-resistant prostate cancer; CRS, cytokine-release syndrome; mCRPC, metastatic castration-resistant prostate cancer; nmHSPC, non-metastatic hormone-sensitive prostate cancer; PSA, prostate-specific antigen; PSMA, prostate-specific membrane antigen; RR, response rate; SC, subcutaneous; TEAE, treatment-emergent adverse event.
Novel BiTE targets
In addition to PSMA-targeting BiTEs, trials have been conducted testing BiTES targeting STEAP1, PSCA, hK 2 and TMEFF2.
STEAP1 is a metalloreductase protein highly expressed in PCa metastases, 110 which contributes to PCa growth via epithelial-to-mesenchymal transformation and cell proliferation. The antigen is specific for malignant cells with low expression in healthy tissues. 111 STEAP1 was evaluated as a therapeutic target in PCa through the development of Xaluritamig, a novel TCE with two identical target anti-STEAP1 antigen-binding domains and an effector anti-CD3 domain. Following promising efficacy and tolerability in monotherapy phase I dose escalation, 112 a randomised dose-optimisation study of 95 patients with heavily pretreated mCRPC was conducted. PSA50 responses were achieved in 50% of patients and 28% had PSA90 responses across all intravenous dose levels (0.75 mg weekly, 1.5 mg weekly and 1.5 mg q-2 weekly). Serious TRAEs occurred in 60% of patients across all cohorts, most commonly musculoskeletal (24%), cytokine-release syndrome (CRS; 20%) and infection (9%). The rate of musculoskeletal inflammatory TRAEs was lowest in the 1.5 mg q-2 weekly cohort. CRS was generally considered manageable, with only 9.4% of patients suffering grade 3 CRS, and no grade 4/5 CRS events. Step-dosing to a target dose of 1.5 mg q-2 weekly was the most favourable balance of efficacy and safety and has been selected as the recommended dose for the recently commenced phase III clinical trial. 113
PSCA is a cell-cycle regulatory protein whose function is incompletely characterised. 114 Whilst PSCA mRNA is non-specifically expressed in healthy and malignant prostate cells, translation and expression of the PSCA protein is more specific to PCa, particularly in the presence of castration resistance and bony metastases. 115 PSCA was evaluated as a therapeutic target in a phase I trial of the PSCA-targeting BiTE GEM3PSCA in patients with PSCA-expressing advanced cancer failing standard therapy, including patients with PCa. The trial was terminated, and to our knowledge, no results have been publicly released.
hK2 is a serine protease that contributes to the activation of complex downstream oncogenic processes. It is structurally related to PSA and is specifically expressed in PCa cells. 116 An ongoing phase I trial is evaluating JNJ-78278343, a hK-2-CD3 BiTE, combined with JNJ-87189401, a PSMA-CD28 BiTE, in patients with mCRPC. The dual activation of CD3 and CD28 costimulatory pathways is hypothesised to produce a potentiated T-cell response. Results are awaited. 117
TMEFF2 is a transmembrane protein involved in androgen receptor regulation, which is highly and specifically expressed throughout all PCa disease stages.118,119 A recently published phase I trial reported results for 82 patients with mCRPC refractory to chemotherapy treated with JNJ-902, a TMEFF2-CD3 BiTE. Across nine dose levels, 12% of patients achieved a PSA50 response, though a dose-response relationship was not observed and a recommended dose for phase II was not determined. 120
A summary of novel BiTEs targeting these TAAs is presented in Table 3.
Novel PCa BiTE trials with published results.
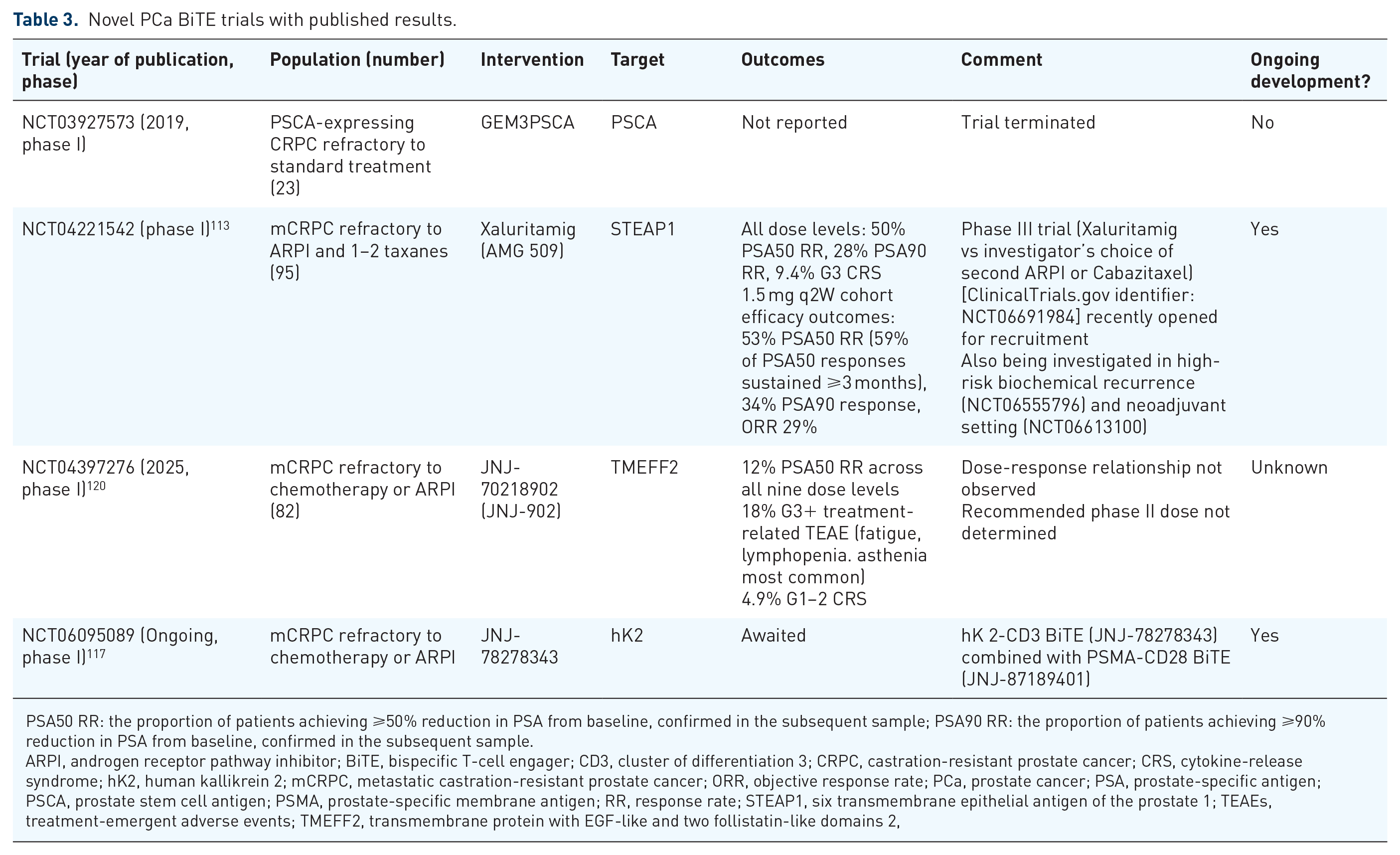
PSA50 RR: the proportion of patients achieving ⩾50% reduction in PSA from baseline, confirmed in the subsequent sample; PSA90 RR: the proportion of patients achieving ⩾90% reduction in PSA from baseline, confirmed in the subsequent sample.
ARPI, androgen receptor pathway inhibitor; BiTE, bispecific T-cell engager; CD3, cluster of differentiation 3; CRPC, castration-resistant prostate cancer; CRS, cytokine-release syndrome; hK2, human kallikrein 2; mCRPC, metastatic castration-resistant prostate cancer; ORR, objective response rate; PCa, prostate cancer; PSA, prostate-specific antigen; PSCA, prostate stem cell antigen; PSMA, prostate-specific membrane antigen; RR, response rate; STEAP1, six transmembrane epithelial antigen of the prostate 1; TEAEs, treatment-emergent adverse events; TMEFF2, transmembrane protein with EGF-like and two follistatin-like domains 2,
CAR-T cells
CAR-T cell therapy represents a promising approach to target PCa TAAs. Current CAR-T cell platforms rely on the ex vivo enrichment and engineering of patient-derived T cells, incorporating strategies to enhance tumour infiltration and persistence. Most CAR-T cell clinical trials remain at the phase I stage, assessing safety and dose-limiting toxicities. Whilst early efficacy and tolerability are promising, serious adverse events highlight the need for dose-optimisation and better delivery approaches to ensure safety and effectiveness.
Similar to BiTEs, CAR-T cells have been engineered to target common PCa TAAs such as PSMA, PSCA, STEAP1 and six transmembrane epithelial antigens of the prostate 2 (STEAP2).121–125 Newer generation CARs have been designed to target more bespoke antigens, including Lewis Y (LeY), Mucin-1 (MUC-1), EphA2 and B7-H3.126–129 Other highly expressed cell surface molecules, like DLL3 and carcinoembryonic antigen-related cell adhesion molecule 5 (CEACAM5), are promising neuroendocrine tumour antigens.130–132
CAR-T cell manufacturing involves isolating, activating and expanding T cells. Activated T cells are genetically modified to express a CAR construct using lentiviral, retroviral or non-viral methods like electroporation or CRISPR.133,134 The CAR is a synthetic receptor that combines an extracellular domain for TAA recognition with intracellular signalling domains like 4-1BB or CD28 to activate T cells upon antigen binding. The specific intracellular co-stimulatory signalling domain is critical in determining a CAR’s sensitivity for tumour antigen expression. 135
Advancements in CAR-T cell engineering
CAR-T cell engineering has evolved through five generations, with the latest advancements improving efficacy, overcoming tumour-induced immunosuppression, and enhancing persistence in the hostile TIME. 136 Key innovations include CAR-T cells, which deliver cytokines, constructs designed to resist immunosuppressive signals, tandem and bispecific CARs for broader targeting, and memory-enriched cells for improved persistence. In addition, switchable and universal CARs enhance specificity and safety, contributing to better therapeutic outcomes.
TRUCKs (T-cell redirected universal cytokine-mediated killing) enhance tumour infiltration by delivering or inducing pro-inflammatory cytokines like IL-12 and IL-15, which promote T-cell proliferation, activation and survival in solid tumours.137–139 IL-12- or IL-15-expressing CAR-T cells can also stimulate endogenous T and NK cells, whilst downstream mediators such as IFNγ activate macrophages and dendritic cells, further amplifying innate and adaptive immune responses.140,141
Additional CAR-T modifications target immunosuppressive factors in the TIME, such as TGF-β and PD-L1.122,142,143 Strategies include dominant-negative or decoy receptors that neutralise these suppressive molecules and engineered PD-1/CD28 switches that convert inhibitory signals into activating ones. 144 One example is the development of TGF-β receptor–modified (TGF-βRIIDN) CAR-T cells targeting PSMA or STEAP2 in PCa, which have demonstrated enhanced anti-tumour efficacy and increased persistence in PCa cell lines and LuCAP PDX models.125,142
Newer CAR constructs incorporate programmable payloads like cytokines, chemokines or BiTEs, enhancing tumour targeting whilst reducing toxicity.145–147 Strategies such as chemokine receptors and tandem or bispecific CARs improve tumour homing and broaden targeting. 148 Notable examples include a PSMA/NKG2DL tandem CAR and MUC1/PSCA CAR-T cells combined with anti-PD-1, both showing strong preclinical activity.149,150
CAR-T cells with enhanced memory traits and self-renewal ability have been engineered to boost their persistence in solid tumours. Human memory T cells include stem-like CD8+ memory T-cell progenitors, which can differentiate into either functional stem-like T (TSTEM) cells or dysfunctional T progenitor exhausted (TPEX) cells. Enriching TSTEM cells boosts the expression of genes involved in cell replication pathways, thereby enhancing proliferative capacity and persistence in vivo. 151 Efforts to boost metabolic fitness include overexpressing transcription factors like FOXO1 to promote a stem-like phenotype and enhance CAR-T cell efficacy against solid tumours.152,153
Advanced CAR-T modifications include ‘logic-gated’ systems that enhance specificity by requiring recognition of multiple antigens, reducing off-target toxicity. 154 Switchable and universal CARs offer flexible targeting, with platforms like UniCAR and RevCAR directing CAR-T cells to PSCA, PSMA and other targets.155–157 A PSMA-specific compound paired with switchable CAR-T cells shows promise for clinical translation. 158
Collectively, these preclinical CAR-T innovations will lay the foundation for safer and more effective therapies for clinical translation.
Current clinical trial outcomes
As technological advances progress, multiple phase I CAR-T trials have been simultaneously conducted in mCRPC. These studies have primarily targeted PSCA and PSMA, with ongoing studies evaluating therapies directed at STEAP1 and STEAP2 (Table 4), commonly involving lymphodepletion regimens consisting of cyclophosphamide (Cy) or Cy/fludarabine (Cy/Flu).
CAR-T cell trials in prostate cancer with published results.

PSA30 RR: the proportion of patients achieving ⩾30% reduction in PSA from baseline, confirmed in the subsequent sample; PSA50 RR: the proportion of patients achieving ⩾50% reduction in PSA from baseline, confirmed in the subsequent sample.
CAR-T, chimeric antigen receptor-T; CRS, cytokine-release syndrome; DLT, dose-limiting toxicity; hK2, human kallikrein 2; mCRPC, metastatic castration-resistant prostate cancer; PSA, prostate-specific antigen; PSCA, prostate stem cell antigen; PSMA, prostate-specific membrane antigen; RR, response rate; STEAP2, six transmembrane epithelial antigen of the prostate 2; TEAE, treatment-emergent adverse event; TSCM, stem cell memory T cell;
A phase I trial of PSCA-directed CAR-T cells in mCRPC patients showed encouraging results, with 4 of 14 patients achieving a ⩾30% reduction in PSA levels and radiographic improvements. 123 However, limited CAR-T cell persistence beyond 28 days highlighted the need for dosing and combination strategy optimisation. A favourable toxicity profile was reported, with CRS occurring in 36% of participants, leading to the ongoing phase Ib trial with or without radiation and outpatient dosing. 163
A phase I trial of BPX-601, an autologous PSCA-directed GoCAR-T® product, showed a PSA50 response in 56% of patients, with 44% achieving PSA90 responses. BPX-601 cells persisted in patients’ peripheral blood for up to 250 days. However, the trial was terminated due to two dose-limiting toxicities and two treatment-related deaths, without a recommended phase II dose. Despite these challenges, cytokine and chemokine induction linked to IFN-γ-driven antitumor activity was observed. 159
Another phase I trial of PCa-directed CAR-T cells engineered with a dominant-negative TGF-β receptor (CART-PSMA-TGFβRDN) showed feasibility and generally safe application, with three patients achieving a PSA reduction of ⩾30%. However, five patients developed grade ⩾2 CRS, including one who experienced a >98% PSA reduction but died from grade 4 CRS and sepsis. These findings highlight the need for further optimisation of TGF-β-resistant CAR-T cells in terms of safety and efficacy. 122
Overall, clinical trial results confirm that current-generation CAR-T cells are feasible and safe in patients, with later-generation constructs and combination therapies poised to significantly enhance outcomes.
Future of CAR-T cell therapies
Whilst clinical trials show promise, several challenges remain in enhancing CAR-T cell therapy for PCa. Key areas for improvement include increasing CAR-T cell infiltration, boosting persistence and activation and minimising toxicities. Moreover, antigen diversity may necessitate personalised strategies and careful patient selection to address these challenges.
The immunosuppressive TIME, which limits CAR-T efficacy, is being addressed through CAR-T designs, combination therapies, and agents like ICIs, chemotherapies and VEGF or TGF-β targeting drugs.126,164–167 Pre-treatment with carboplatin enhanced LeY CAR-T efficacy in PCa PDXs by inducing pro-inflammatory changes in the TME, improving CAR-T infiltration and activation, highlighting the potential of TIME-modulating agents to boost CAR-T therapy. 126
Patient toxicity and the high cost of CAR-T therapies remain major hurdles. Currently, CAR-T therapy uses autologous T cells but advances in mRNA-LNPs and CRISPR are enabling in-body CAR-T cell engineering, eliminating the need for ex vivo manipulation.168–170 In addition, the generation of allogeneic, or off-the-shelf CAR-T cells is being explored through the knockout of endogenous T-cell receptors (TCRs), preventing graft-versus-host disease whilst preserving CAR function.171,172 These approaches could reduce the logistical and cost challenges of autologous therapies, improving CAR-T safety and efficacy whilst making it more accessible to a broader patient population.
Cost-effectiveness
The cost of bespoke immunotherapy treatments can be extremely high; for example, the first commercially available CAR-T product Tisagenleucel carried an upfront cost of US $475,000 when the product was brought to market in 2017 for potential treatment of B-cell acute lymphoblastic leukaemia (B-ALL). 173 In comparison, newer generation products used in PCa may be associated with both lower cost and reduced efficacy. BiTEs are generally associated with lower ‘off-the-shelf’ production costs than CAR-T cells. 174 Blinatumomab for curative treatment of B-ALL and Tarlatamab for palliative treatment of extensive-stage small cell lung cancer provide precedent for the commercialisation of BiTEs across a more diverse range of disease settings and treatment intentions.
As these immunotherapies move into phase III clinical trials against standard-of-care drugs, health economic analysis will be crucial to assessing the potential benefit. Furthermore, economic analyses of new immunotherapeutics for PCa may face difficulties in comparison with on-off treatments with the theoretic potential of persistent treatment effects (such as CAR-T cells and therapeutic cancer vaccines) versus indefinite palliative oral therapy (such as ARPIs). The former class of therapy may appear incrementally more cost-effective with analyses conducted at later time points, whilst indefinite palliative oral therapy may appear incrementally less cost-effective over time.
Summary timeline
A timeline summarising key events in the development of immunotherapy for PCa is presented in Figure 3.

Timeline of key immunotherapy trials and drugs.
Conclusion
After many years of disappointing results with immunotherapy in metastatic PCa, TCEs have shown promising activity in mCRPC. Despite the success of ICIs in many solid organ tumours, these drugs alone or in combination with other systemic therapies do not improve efficacy outcomes for mCRPC. Whilst there may be a role for ICIs for patients with MMR deficient or high TMB tumours, the vast majority of patients have a ‘cold’ immunological phenotype, and ICIs have proven ineffective. Whilst early experience with TCEs was limited by issues with drug delivery, neutralisation and toxicity; data for newer agents are more promising and the class is expected to continue to progress, exemplified by the STEAP1-targeted TCE Xaluritamig, which is being taken forward to a phase III clinical trial. Similarly, CAR-T cells represent a promising approach to therapy, with their own limitations of toxicity and high cost. Ongoing drug development seeks to refine these agents, and it is likely that personalised approaches and consideration of combination therapies will be of interest.