
Abstract
Background:
Chemotherapy-induced thrombocytopenia (CIT) increases the risk of bleeding, necessitates chemotherapy dose reductions and delays, and negatively impacts prognosis.
Objectives:
This study aimed to evaluate the efficacy and safety of hetrombopag for the management of CIT in patients with advanced solid tumors.
Design:
A multicenter, randomized, double-blind, placebo-controlled, phase II study.
Methods:
Patients with advanced solid tumors who experienced a chemotherapy delay of ⩾7 days due to thrombocytopenia (platelet count <75 × 109/L) were randomly assigned (1:1) to receive oral hetrombopag at an initial dose of 7.5 mg once daily or a matching placebo. The primary endpoint was the proportion of treatment responders, defined as patients resuming chemotherapy within 14 days (platelet count ⩾100 × 109/L) and not requiring a chemotherapy dose reduction of ⩾15% or a delay of ⩾4 days or rescue therapy for two consecutive cycles.
Results:
Between 9 October 2021 and 5 May 2022, 60 patients were randomized, with 59 receiving ⩾1 dose of assigned treatment (hetrombopag/placebo arm, n = 28/31). The proportion of treatment responders was significantly higher in the hetrombopag arm than in the placebo arm [60.7% (17/28) versus 12.9% (4/31); difference of proportion: 47.6% (95% confidence interval (CI): 26.0–69.3); odds ratio = 10.44 (95% CI: 2.82–38.65); p value (nominal) based on the Cochran–Mantel–Haenszel: <0.001)]. During the double-blind treatment period, grade 3 or higher adverse events (AEs) occurred in 35.7% (10/28) of patients with hetrombopag and 38.7% (12/31) of patients on placebo. The most common grade 3 or higher AEs were decreased neutrophil count [35.7% (10/28) versus 35.5% (11/31)] and decreased white blood cell count [17.9% (5/28) versus 19.4% (6/31)]. Serious AEs were reported in 3.6% (1/28) of patients with hetrombopag and 9.7% (3/31) of patients with placebo.
Conclusion:
Hetrombopag is an effective and well-tolerated alternative for managing CIT in patients with solid tumors.
Trial registration:
ClinicalTrials.gov identifier: NCT03976882.
Keywords
Introduction
Chemotherapy-induced thrombocytopenia (CIT) is a frequent complication of myelosuppressive chemotherapy. 1 Patients with cancer are at a heightened risk of bleeding due to antitumor treatments such as chemotherapy, radiation, and anti-angiogenic therapy, as well as from tumor invasion. This risk is exacerbated by thrombocytopenia. Moreover, patients with CIT who suffer from major bleeding events are associated with a poor clinical prognosis. 2
Currently, the treatment options for CIT include platelet transfusions, medications, and adjustments to chemotherapy dosages or schedules. However, platelet transfusions are scarce and have limited efficacy. Delays in chemotherapy, as well as dose reductions or discontinuations, can lead to a decrease in the relative dose intensity (RDI) of chemotherapy, 3 resulting in unfavorable outcomes.4–8 Although recombinant human interleukin-11 (rhIL-11) has been approved for the management of CIT, 9 its clinical use is restricted by adverse effects such as arrhythmias, fluid retention, and pulmonary edema, and by limited effectiveness.9,10 Therefore, the lack of a safe and effective therapeutic approach continues to represent an unmet clinical need in CIT management.
Recent studies have highlighted the potential of targeting the thrombopoietin (TPO)/thrombopoietin receptor (TPO-R) pathway as a promising intervention for CIT. First-generation thrombopoietic agents, including recombinant human TPO (rhTPO) and a pegylated variant known as recombinant human megakaryocyte growth and development factor (PEG-rHuMGDF or MGDF), 11 have proven effective in increasing platelet counts across various clinical scenarios. 12 However, the development of neutralizing antibodies against MGDF, which can result in persistent thrombocytopenia, has been observed in some individuals, leading to the termination of their clinical development. 13 However, the development of these agents was halted due to the emergence of neutralizing antibodies against MGDF in some individuals, which led to persistent thrombocytopenia. Second-generation thrombopoietic agents exert biological effects by binding to and activating the TPO-R, also known as thrombopoietin receptor agonists (TPO-RAs). TPO-RAs include the peptibody romiplostim and the small molecule agents such as eltrombopag, avatrombopag, lusutrombopag, and hetrombopag. 3 Initially approved for treating immune thrombocytopenia (ITP), aplastic anemia (AA), and periprocedural thrombocytopenia in patients with chronic liver disease, these agents present a promising novel option for managing CIT in patients with solid tumors.3,14 Currently, several of these agents are under investigation to explore their potential application in CIT management.3,12,14–17
Hetrombopag (Hengqu®, Jiangsu Hengrui Pharmaceuticals Co., Ltd.), an orally administered, small molecule synthetic TPO-RA, is being developed by Jiangsu Hengrui Pharmaceutical. Hetrombopag is produced by making structural modifications to eltrombopag, which serve to improve its potency and reduce its potential for toxicity.18,19 Hetrombopag received its initial approval in China on 16 June 2021, 20 for treating primary ITP in adult patients who have not responded well to treatments like glucocorticoids and immunoglobulins. 21 In addition, it received conditional approval for treating severe AA in patients who are refractory to immunosuppressive therapy. 22 Meanwhile, hetrombopag is being investigated as a management option for CIT. This randomized, multicenter, placebo-controlled phase II trial is the first to evaluate the efficacy and safety of hetrombopag for the management of CIT in patients with advanced solid tumors.
Methods
Study design and patients
This multicenter phase II study (NCT03976882) of hetrombopag for the treatment of CIT in patients with advanced solid tumors was conducted at 18 sites in China (Supplemental Table 1), including a randomized, double-blind, placebo-controlled core cohort and an open-label, single-arm exploratory cohort (Supplemental Figure 1). The core cohort consisted of a randomized, double-blind, placebo-controlled treatment period (correction phase and two 3-week on-study chemotherapy cycles), followed by an optional open-label extension treatment period (up to four 3-week chemotherapy cycles). Herein, we report the results from the double-blind treatment period in this phase II study.
Eligible patients included adults aged 18–75 years with histologically or cytologically confirmed advanced solid tumors, including but not limited to breast cancer, bladder cancer, small-cell lung carcinoma, and non-small-cell lung carcinoma (NSCLC), receiving chemotherapy every 3 weeks. Their planned chemotherapy cycle had been delayed for at least 1 week due to thrombocytopenia (platelet count <75 × 109/L). Chemotherapy regimens included at least one of the following: antimetabolites such as gemcitabine, platinum-based agents like carboplatin, nedaplatin, cisplatin, and lobaplatin, an anthracycline (e.g. doxorubicin, daunorubicin, epirubicin), or an alkylating agent (e.g. cyclophosphamide, ifosfamide). All patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0–1, a life expectancy of more than 12 weeks at screening, and were able to receive at least two additional cycles of their current chemotherapy regimen. Key exclusion criteria included any hematological disorders other than CIT (such as leukemia, primary ITP, myeloproliferative neoplasms, multiple myeloma, and myelodysplastic syndrome); thrombocytopenia resulting from causes other than CIT (such as chronic liver disease, splenomegaly, infection, and bleeding); a history of severe cardiovascular disease, any arterial or venous thrombosis within the preceding 6 months; severe hemorrhage within 2 weeks before screening; and previous treatments with TPO-R agonists (e.g. eltrombopag, romiplostim), rhTPO, or rhIL-11 within 30 days before screening. The full inclusion and exclusion criteria are detailed in Supplemental Table 2.
The trial was conducted in accordance with the Declaration of Helsinki and the Good Clinical Practice Guidelines. The protocol and all amendments were approved by the institutional review board or independent ethics committee of each participating clinical center (Supplemental Table 1). All patients provided written informed consent before study procedures.
Treatment interventions
After a screening period of less than 4 weeks, patients were randomized (in a 1:1 ratio) to receive hetrombopag or a matching placebo, starting at a dose of 7.5 mg once daily, stratified by baseline platelet count (<50 × 109/L or ⩾50 × 109/L), for the entire double-blind treatment period or until withdrawal from the study, based on a computer-generated randomization code generated by an interactive web response system. The color, shape, size, and texture of the hetrombopag and placebo tablets were identical. All personnel involved in the study conduct and interpretation (including investigators, coordinators, data collectors, and sponsor staff) were blinded to the treatment group assignment (though not to dose levels), and patients were also blinded to their treatment. Central randomization data were kept strictly confidential until unblinding.
The initial dose of hetrombopag/placebo was set at 7.5 mg once daily, which could be titrated to maintain platelet counts, up to a maximum of 15 mg per day (rationale for dose selection is detailed in Supplemental File 1). To ensure efficacy and safety, dose titration was recommended to be maintained for 2 weeks each time. This titration approach empowered patients to tailor their dosage based on platelet levels, thereby achieving personalized and optimal treatment outcomes. The principles of dose titration for hetrombopag/placebo in the management of CIT in advanced solid tumor patients are presented in Supplemental Table 3.
Following the double-blind treatment period, patients may participate in an optional open-label extension treatment period for up to four chemotherapy cycles, at the discretion of the investigators. Patients who received hetrombopag during the double-blind treatment period continued their therapy at their current doses, while those who received a placebo were switched to hetrombopag treatment at an initial dose of 7.5 mg once daily. The key criteria for study withdrawal included intolerable toxicities, protocol violations, poor compliance, and withdrawal by investigators.
Efficacy endpoints and assessments
Patients underwent weekly assessments of their platelet counts during the double-blind treatment period. The platelet response was defined as achieving a platelet count of ⩾100 × 109/L. In the initial 14 days of treatment, known as the correction phase, platelet counts were checked weekly, or more frequently if deemed necessary by the investigator. Patients were allowed to resume their first planned chemotherapy cycle once their platelet counts reached ⩾100 × 109/L. If a patient’s platelet count did not meet this threshold after the correction phase, or if a subsequent chemotherapy cycle was delayed by ⩾4 days, the use of rhTPO and rhIL-11 was permitted at the discretion of the investigator to expedite the resumption of chemotherapy.
The primary endpoint was the proportion of treatment responders, defined as patients who could resume their planned chemotherapy with a platelet count of ⩾100 × 109/L within 14 days of the first dose of study treatment, and complete two consecutive planned chemotherapy cycles without any modification to the chemotherapy regimen (i.e. dose reduction of ⩾15%, delay of ⩾4 days, or discontinuation) or the need for rescue therapy due to thrombocytopenia.
Secondary efficacy endpoints included the proportion of patients who achieved a documented platelet response and resumed their first planned chemotherapy cycle within 14 days of initiating study treatment; the time from study treatment initiation to the first documented platelet response and the resumption of the first planned chemotherapy cycle; the proportion of patients who completed two chemotherapy cycles without the need for rescue therapy or dose modifications due to thrombocytopenia; the duration of the platelet response, the nadir of the platelet count, and the duration of severe thrombocytopenia (defined as a platelet count of ⩽50 × 109/L) from the beginning of the first on-study chemotherapy cycle until the end of the double-blind treatment period.
During the double-blind treatment period, exploratory endpoints included the proportion of patients who achieved platelet counts of ⩾100 × 109/L or ⩾75 × 109/L on Day 21 of on-study chemotherapy cycle 1 or cycle 2, the proportion of patients who achieved a platelet count nadir of <75 × 109/L or <50 × 109/L after Day 14 of on-study chemotherapy cycle 1, the proportion of patients who required at least one platelet transfusion or protocol-defined rescue therapy, and the platelet count recorded at every scheduled visit.
Safety assessments
Patients were continuously monitored for adverse events (AEs) until 14 days after their last dose of the study treatment. Vital signs, physical examinations, clinical laboratory evaluations, electrocardiograms, echocardiography, and abdominal ultrasound were performed and recorded at each scheduled visit during the double-blind treatment. AEs were coded to the preferred terms of the Medical Dictionary for Regulatory Activities v22.0 and graded according to the NCI Common Terminology Criteria for Adverse Events, version 5.0. Predefined adverse events of special interest (AESIs) included potential drug-induced liver injuries, thrombosis, and/or thromboembolic events. The incidence and severity of bleeding were recorded and assessed according to the World Health Organization (WHO) bleeding scale (grade 0, no bleeding; grade 1, petechiae; grade 2, mild blood loss; grade 3, gross blood loss; and grade 4, debilitating blood loss).
Statistical analyses
For the core cohort, 50 patients were planned to be enrolled and randomized at a ratio of 1:1. During the double-blind treatment period, the assumed response rate was 80% for the hetrombopag group and 40% for the placebo group. The sample size was designed to provide an 85% power to detect this difference at a one-sided significance level of 0.025. In addition, if the response rate in the hetrombopag group were assumed to be 70%, the power to detect a difference between the two groups would be approximately 57%. Efficacy was assessed in the full analysis set, which included all randomized patients who received at least one dose of the assigned study treatment. The safety set comprised all enrolled patients who received at least one dose of the study treatment. The primary endpoint and secondary binary efficacy endpoints comparison between hetrombopag and placebo were evaluated using the Cochran–Mantel–Haenszel (CMH) test, which was adjusted for baseline platelet count (<50 × 109/L or ⩾50 × 109/L). Non-completers were considered as failures, and missing data were imputed using the non-completer failure (NCF) method. The odds ratio (OR) and 95% confidence interval (CI) were also provided. Prespecified subgroup analyses were conducted for the primary endpoint based on age (<60 or ⩾60 years), the number of chemotherapy agents used (1 or ⩾2), and baseline platelet count (<50 × 109/L or ⩾50 × 109/L). The median time interval from study treatment initiation to the first documented platelet response was estimated using the Kaplan–Meier method (post hoc analysis). Descriptive statistical analyses were employed to summarize other secondary and exploratory endpoints using the NCF method or observed cases approach. Results were presented as the number (percentage), mean ± standard deviation), or median (Min, Max). Data analyses were performed using SAS software (version 9.4, SAS Institute Inc, Cary, USA).
Reporting guideline
The reporting of this study conforms to the Consolidated Standards of Reporting Trials (CONSORT) 2010 Statement 23 (Supplemental File 2).
Results
Patient characteristics
Between 9 October 2021 and 5 May 2022, 101 patients were screened for eligibility, of whom 60 were enrolled and randomly assigned (1:1) to either the hetrombopag (n = 29) or placebo (n = 31) study groups (Figure 1). One patient in the hetrombopag group did not receive the assigned treatment due to the withdrawal of informed consent. As of the data cutoff date (23 February 2023), 22 patients (75.9%) in the hetrombopag group and 26 patients (83.9%) in the placebo group had completed the double-blind treatment period. A total of 11 patients (18.6%) discontinued the study treatment permanently, primarily because of patient withdrawal [3 (10.3%) in the hetrombopag group and 3 (9.7%) in the placebo group] and physician decisions [2 (6.9%) in the hetrombopag group and 1 (3.2%) in the placebo group].

Study profile.
The median age of patients in the core cohort was 58 years, with 31 (52.5%) males and 28 females (47.5%). The majority of patients (71.2%, 42/59) had an ECOG score of 1. A total of 44 (74.6%) patients had a baseline platelet count of ⩾50 × 109/L. Forty-one (69.5%) patients had received systemic treatment for advanced/metastatic disease; of these, 27 (27/41; 65.9%) received first-line systemic therapy. Fourteen (50.0%) patients in the hetrombopag group and 13 (41.9%) in the placebo group received chemotherapy combined with immunotherapy or targeted therapy. Patient characteristics were generally well balanced between the hetrombopag and placebo groups at baseline (Table 1).
Baseline characteristics.

In the hetrombopag group, one patient each had endometrial, cervical, breast, nasopharyngeal carcinoma, and malignant melanoma. For the placebo group, there was also one patient each with malignant melanoma, germ cell tumor, gallbladder cancer, and NSCLC.
ECOG, Eastern Cooperative Oncology Group; Min, minimum; Max, maximum; NSCLC, non-small-cell lung cancer; SD, standard deviation.
Primary endpoint
As shown in Figure 2(a), the hetrombopag group exhibited a higher proportion of treatment responders (60.7%; 17/28) compared to the placebo group (12.9%; 4/31), with a difference in proportion of 47.6% (95% CI: 26.0–69.3). The OR for hetrombopag versus placebo was 10.44 (95% CI: 2.82–38.65; nominal p value based on the CMH test: <0.001).

(a) Proportion of treatment responders during the double-blind treatment period (primary endpoint). (b) Proportions of patients who achieved their first documented platelet response during the correction phase.
Subgroup analysis revealed that the proportion of treatment responders was consistently higher in patients receiving hetrombopag than in those receiving placebo across all prespecified subgroups (Supplemental Table 4).
Efficacy during the correction phase
A higher proportion of patients in the hetrombopag group achieved a documented platelet response and resumed the first planned chemotherapy cycle within 14 days of study treatment initiation compared to the placebo group (85.7% [24/28] versus 48.4% [15/31]); difference of proportion: 37.2% (95% CI: 15.2–59.2); OR = 6.21 (95% CI: 1.69–22.89); nominal p = 0.003; Table 2). During the correction phase, the median time from study treatment initiation to the first documented platelet response was 7.5 days in the hetrombopag group, compared to 13.0 days in the placebo group [post hoc analysis; Figure 2(b)].
Secondary efficacy endpoints during the double-blind treatment period.

Number of patients analyzed using the NCF approach
Hetrombopag versus placebo.
The binary efficacy endpoints were compared between the hetrombopag and placebo groups using Cochran–Mantel–Haenszel tests, stratified by baseline platelet count (<50 × 109/L or ⩾50 × 109/L), and the corresponding OR and 95% CI values were calculated. All displayed p values for secondary endpoints are nominal p values.
Number of patients analyzed using the observed cases approach.
Since the start of the first on-study chemotherapy cycle through the end of the double-blind treatment period.
CI, confidence interval; Min, minimum; Max, maximum; NCF, non-completer failure; OR, odd ratio.
Efficacy during the maintenance phase
The proportion of patients who completed two chemotherapy cycles without requiring rescue therapy or dose modifications due to thrombocytopenia was significantly higher in the hetrombopag group (60.7%; 17/28) compared to the placebo group (25.8%; 8/31) [difference of proportion: 34.7% (95% CI: 11.7–57.7); hetrombopag versus placebo: OR = 4.96 (95% CI: 1.55–15.89); nominal p value: 0.006; Table 2].
The proportion of patients who achieved platelet counts of ⩾100 × 109/L or ⩾75 × 109/L on Day 21 of on-study chemotherapy cycles 1 and 2, as well as the proportion of patients who reached a platelet count nadir of <75 × 109/L or <50 × 109/L after Day 14 of on-study chemotherapy cycle 1, are detailed in Supplemental Tables 5 and 6. In the core cohort, 3 (15.8%; 3/19) patients in the hetrombopag group and 14 (53.8%; 14/26) patients in the placebo group were observed to experience a platelet count nadir of <50 × 109/L after Day 14 of on-study chemotherapy cycle 1. Furthermore, 18 (81.8%; 18/22) patients in the hetrombopag group and 8 (30.8%; 8/26) patients in the placebo group achieved platelet counts of ⩾75 × 109/L on Day 21 of the second chemotherapy cycle.
Usage of rescue therapy during the double-blind treatment period
During the double-blind treatment period, 5 (17.9%; 5/28) patients in the hetrombopag group and 19 (61.3%; 19/31) patients in the placebo group received at least one platelet transfusion or protocol-defined rescue therapy (rhTPO and rhIL-11; Supplemental Table 7).
Platelet count during the double-blind treatment period
Platelet counts during the double-blind treatment period are illustrated in Figure 3. The mean platelet counts of patients in the hetrombopag group were consistently higher than those in the placebo group, which maintained levels between 100.0 and 200.0 × 109/L.

Platelet counts during the double-blind treatment period.
As shown in Table 2, patients in the hetrombopag group had a median duration of platelet response of 28.0 days, a median platelet count nadir of 79.0 × 109/L, and a median duration of severe thrombocytopenia of 12.0 days from the start of the first on-study chemotherapy cycle to the end of the double-blind treatment period. By contrast, patients in the placebo group experienced a median duration of platelet response of 14.0 days, a median platelet count nadir of 51.0 × 109/L, and a median time duration of severe thrombocytopenia of 10.5 days during the same period.
Safety
The median duration of treatment exposure during the double-blind treatment period was 51.0 days in the hetrombopag group and 62.0 days in the placebo group. Only one patient (3.2%) in the placebo group reported an AE that resulted in dose interruption (venous thrombosis limb), while no such AEs were reported in the hetrombopag group. In addition, there were no AEs leading to study withdrawal or dose discontinuation of the study treatment in either treatment arm (Table 3). In the hetrombopag group, 9 patients (31.2%) experienced dose escalation, while 21 patients (67.7%) in the placebo group underwent such adjustments. In addition, 12 patients (42.6%) in the hetrombopag group and 2 patients (6.5%) in the placebo group experienced dose reductions.
Adverse events during the double-blind treatment period.
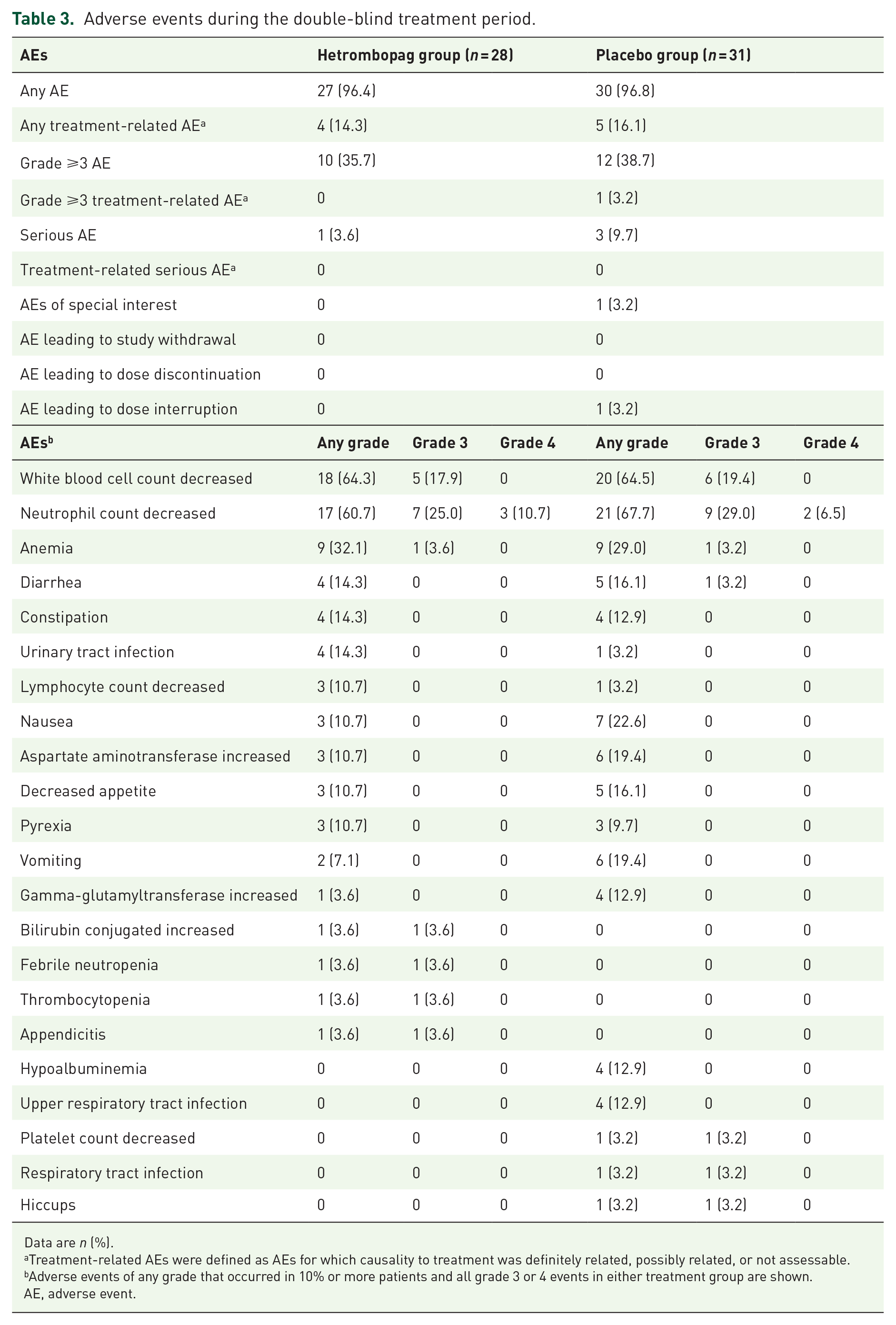
Data are n (%).
Treatment-related AEs were defined as AEs for which causality to treatment was definitely related, possibly related, or not assessable.
Adverse events of any grade that occurred in 10% or more patients and all grade 3 or 4 events in either treatment group are shown.
AE, adverse event.
Among the 59 patients in the safety analysis set, AEs of any grade occurred in 27 (96.4%) patients in the hetrombopag group and 30 (96.8%) patients in the placebo group. The most common AEs were decreased white blood cell count (64.3% in the hetrombopag group versus 64.5% in the placebo group), decreased neutrophil count (60.7% versus 67.7%), and anemia (32.1% versus 29.0%; Table 3). Four patients (14.3%) in the hetrombopag group and five patients (16.1%) in the placebo group experienced treatment-related AEs (Supplemental Table 8). Grade 3 or 4 AEs, most of which were decreased neutrophil count (35.7% in the hetrombopag versus 35.5% in the placebo group) and decreased white blood cell count (17.9% versus 19.4%; Table 3), were reported in 10 (35.7%) patients receiving hetrombopag and 12 (38.7%) patients receiving placebo; of these, only one event (grade 3 decreased neutrophil count) in the placebo group was considered by the investigator to be related to the study treatment.
Serious AEs occurred in one patient (3.6%) in the hetrombopag group (febrile neutropenia and thrombocytopenia) and three patients (9.7%) in the placebo group (one with pneumonia, one with hiccups, and one with respiratory tract infection and venous thrombosis limb); however, none of these SAEs were considered to be related to study treatment (Supplemental Table 9). No deaths regardless of cause occurred in either treatment group.
During the double-blind treatment period, only one patient (3.2%) in the placebo group experienced an AESI (venous thrombosis limb); meanwhile, no patients treated with hetrombopag reported any AESIs. No patient experienced bleeding symptoms with a WHO score of ⩾2 during this period.
Discussion
This phase II study is the first trial to evaluate the efficacy and safety of hetrombopag in managing CIT in patients with advanced solid tumors. During the double-blind treatment period, prominent improvements were observed in the primary endpoint and all secondary endpoints for patients treated with hetrombopag at an initial dose of 7.5 mg once daily, compared to those receiving a placebo. Furthermore, the AE profile of hetrombopag was comparable to that of placebo, with no increased incidence of thromboembolic events, confirming its safety in this patient population.
The primary endpoint of this study was the proportion of treatment responders, which referred to patients who could resume their planned chemotherapy within 14 days of the first dose of study treatment and complete two consecutive planned chemotherapy cycles without any modification to the chemotherapy regimen or the need for rescue therapy. We found that hetrombopag increased the proportion of treatment responders compared to placebo. The improvement was consistent across all prespecified subgroups, suggesting that hetrombopag could offer stable clinical benefits to a diverse patient population. Several pivotal phase II and III studies have demonstrated the efficacy of romiplostim, eltrombopag, and avatrombopag in managing or preventing CIT. 3 For instance, a phase II study comparing romiplostim with untreated observation in patients with CIT found that 14 of 15 romiplostim-treated patients achieved the primary endpoint of platelet count correction to >100 × 109/L within 3 weeks, compared to only one of eight in the untreated group. 17 Similarly, a phase II study of eltrombopag showed that treated patients had higher platelet counts, fewer occurrences of grade 3 or 4 CIT, more rapid platelet count recovery, and fewer dose reductions or treatment delays due to thrombocytopenia. 16 However, a phase III study of avatrombopag, while confirming increased platelet counts in treated patients, revealed an unexpectedly strong rebound in platelet counts in the placebo group prior to chemotherapy, with few patients requiring platelet transfusions or chemotherapy dose modifications. It is important to note that this study excluded patients with grade 2 or higher CIT (other than from the current chemotherapy regimen) within 6 months of screening and those who had received more than two previous lines of chemotherapy. In addition, the study focused only on patients with isolated, nadir thrombocytopenia and did not separately assess those with more severe or persistent CIT. 15
During the correction phase, the hetrombopag group exhibited a higher proportion of patients who achieved a documented platelet response and resumed the first planned chemotherapy cycle within 14 days of study treatment initiation, compared to the placebo group. During the maintenance phase, the hetrombopag group demonstrated a higher proportion of patients who completed two consecutive chemotherapy cycles without requiring dose modifications or rescue therapy due to thrombocytopenia, compared to the placebo group. In addition, a greater number of patients in the hetrombopag group achieved platelet counts of ⩾75 × 109/L on Day 21 of the second on-study chemotherapy cycle, compared to the placebo group. As shown in Figure 3, continuous administration of hetrombopag maintained a safe and stable platelet count level (100–200 × 109/L) during two consecutive on-study chemotherapy cycles. Furthermore, only 17.9% of patients in the hetrombopag group required at least one platelet transfusion or protocol-defined rescue therapy during the double-blind treatment period, compared to 61.3% of patients in the placebo group. These findings support the use of hetrombopag at an initial dose of 7.5 mg once daily in advanced solid tumor patients with CIT. Hetrombopag effectively managed CIT by rapidly increasing platelet count levels and allowing patients to resume chemotherapy cycles as planned, thereby facilitating the smooth progression of subsequent chemotherapy cycles. This resulted in a stable platelet response, a low risk of CIT recurrence, and tolerance of the cumulative effects of bone marrow suppression induced by consecutive chemotherapy cycles, ultimately benefiting patients by achieving a desirable chemotherapy RDI. Our findings were comparable with those of a retrospective study conducted by Al-Samkari et al., 24 which evaluated the effectiveness of romiplostim for CIT across four centers. The study’s primary outcome was the attainment of platelet counts ⩾75 × 109/L, with an increase of ⩾30 × 109/L from baseline levels. The study included 173 cancer patients, of which 153 had solid tumors. The results showed that 71% of patients achieved the primary efficacy outcome over a median of four cycles of chemotherapy administered concurrently with romiplostim. In addition, 79% of patients did not require chemotherapy dose reductions or treatment delays, and 89% of patients did not require a platelet transfusion. 24
The AEs reported for hetrombopag were consistent with the safety profile of other TPO-RAs used to manage CIT in patients with advanced solid tumors.15–17,24–33 No new safety signals were identified in this phase II study. Hetrombopag was safe and well tolerated, effectively increasing platelet counts without an increased incidence of thromboembolic events compared to placebo. Currently, there is no evidence of an increased risk of thromboembolism associated with the use of TPO-RAs for managing CIT, although randomized studies have not specifically aimed to assess differences in thromboembolic rates. Previous studies on romiplostim reported a 12-month venous thromboembolism (VTE) rate of 10.2% in a phase II study of 52 patients 17 and 14 VTEs per 100 patient-years in a large observational cohort. 24 Similarly, studies on eltrombopag and avatrombopag have not shown increased thromboembolism rates, with a randomized phase II study of 183 patients showing comparable rates between eltrombopag-treated patients and those who received a placebo (7.3% versus 6.5%), 27 and a randomized phase III study of 122 patients reporting similar rates between avatrombopag-treated and placebo-treated patients (2.4% versus 2.5%). 15 The potential impact of treatment on cancer progression or overall survival cannot be accurately assessed due to the heterogeneity of cancer types and the fact that 45.8% of patients in our phase II study were receiving their first-line systemic therapy. Nevertheless, multiple studies have investigated patient samples across a diverse array of cancer types, including, but not limited to, lung, breast, kidney, colon, ovarian, prostate cancers, sarcomas, and hematologic malignancies.34,35 These studies have consistently shown that the TPO-R c-mpl is expressed at minimal or undetectable levels, which is in stark contrast to the expression levels of other receptors, such as the human epidermal growth factor receptor, erythropoietin receptor, and insulin-like growth factor 1 receptor. These findings strongly suggest that the use of TPO-RAs is unlikely to promote tumor growth in cancer patients. Patients treated with hetrombopag experienced a slightly higher incidence of serious AEs compared to those on placebo; however, none of these events were considered related to the study treatment. In addition, the absence of reported drug-induced liver injuries (another prespecified AESI) and bleeding symptoms with a WHO score of ⩾2 during the double-blind treatment period of this phase II study, potentially due to the limited sample size and brief exposure to hetrombopag, underscored the need for larger, longer-term studies to fully assess the potential risks associated with this agent. Notably, in patients treated with hetrombopag, there were no AEs that led to study withdrawal, dose discontinuation, or interruption, indicating excellent tolerability.
There are a few limitations in the current study that need to be addressed. First, although a decent treatment response was observed during the double-blind treatment period in patients with the initial dose of 7.5 mg hetrombopag, the durability of the treatment response and the safety of hetrombopag need to be further investigated in longer-term studies. Second, future studies could consider including an active comparator in addition to the placebo group to provide a more comprehensive evaluation of the efficacy and safety of hetrombopag.
Conclusion
This phase II study demonstrated that hetrombopag led to a clinically relevant improvement in the proportion of treatment responders among advanced solid tumor patients with CIT, compared to those who received a placebo. Hetrombopag was generally well tolerated, with a manageable safety profile, and an initial dose of 7.5 mg once daily may be appropriate for this patient population. Our findings suggest that hetrombopag could serve as a viable alternative for CIT management in patients with advanced solid tumors, and further investigation is warranted.
Supplemental Material
sj-docx-1-tam-10.1177_17588359241260985 – Supplemental material for Hetrombopag for the management of chemotherapy-induced thrombocytopenia in patients with advanced solid tumors: a multicenter, randomized, double-blind, placebo-controlled, phase II study
Supplemental material, sj-docx-1-tam-10.1177_17588359241260985 for Hetrombopag for the management of chemotherapy-induced thrombocytopenia in patients with advanced solid tumors: a multicenter, randomized, double-blind, placebo-controlled, phase II study by Shukui Qin, Yusheng Wang, Jun Yao, Yanyan Liu, Tienan Yi, Yueyin Pan, Zhendong Chen, Xizhi Zhang, Jin Lu, Junyan Yu, Yanjun Zhang, Peng Cheng, Yong Mao, Jian Zhang, Meiyu Fang, Yanming Zhang, Jing Lv, Runzi Li, Ning Dou, Qian Tang and Jun Ma in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-docx-2-tam-10.1177_17588359241260985 – Supplemental material for Hetrombopag for the management of chemotherapy-induced thrombocytopenia in patients with advanced solid tumors: a multicenter, randomized, double-blind, placebo-controlled, phase II study
Supplemental material, sj-docx-2-tam-10.1177_17588359241260985 for Hetrombopag for the management of chemotherapy-induced thrombocytopenia in patients with advanced solid tumors: a multicenter, randomized, double-blind, placebo-controlled, phase II study by Shukui Qin, Yusheng Wang, Jun Yao, Yanyan Liu, Tienan Yi, Yueyin Pan, Zhendong Chen, Xizhi Zhang, Jin Lu, Junyan Yu, Yanjun Zhang, Peng Cheng, Yong Mao, Jian Zhang, Meiyu Fang, Yanming Zhang, Jing Lv, Runzi Li, Ning Dou, Qian Tang and Jun Ma in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
We are grateful to all patients and their families and all members of the study group. Medical writing support was provided by Lin Dong (PhD, a medical writer at Jiangsu Hengrui Pharmaceuticals Co., Ltd) according to Good Publication Practice Guidelines.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.