
Abstract
MET alterations, including MET exon 14 skipping variants, MET amplification, MET overexpression, and MET fusion, play pivotal roles in primary tumorigenesis and acquired resistance to targeted therapies, especially EGFR tyrosine kinase inhibitors. They represent important diagnostic, prognostic, and predictive biomarkers in many solid tumor types. However, the detection of MET alterations is challenging due to the complexity of MET alterations and the diversity of platform technologies. Therefore, techniques with high sensitivity, specificity, and reliable molecular detection accuracy are needed to overcome such hindrances and aid in biomarker-guided therapies. The current review emphasizes the role of MET alterations as oncogenic drivers in a variety of cancers and their involvement in the development of resistance to targeted therapies. Moreover, our review provides an overview of and recommendations on the selection of various cross-platform technologies for the detection of MET exon 14 skipping variants, MET amplification, MET overexpression, and MET fusion. Furthermore, challenges and hurdles underlying these common detection platforms are discussed.
Introduction
MET (mesenchymal–epithelial transition factor) is a widely expressed tyrosine kinase receptor that binds with the natural ligand hepatocyte growth factor (HGF) and plays a vital role in embryogenesis, cell growth, cell differentiation, and angiogenesis. 1 MET activation negatively affects tyrosine kinase inhibitors (TKIs) effectiveness due to the intertwining between the MET and receptor tyrosine kinase (RTK) [epidermal growth factor receptor (EGFR)] signaling pathways. 2 Dysregulation of the HGF-MET axis potentially arises by a variety of mechanisms, including mutational activation, such as exon 14 splice site alteration, exon 14 ubiquitination site mutation, kinase domain mutation, extracellular domain mutation, and amplification of the MET proto-oncogene or gene copy number (GCN) gain due to polysomy or focal amplification 3 or by overexpression that occurs either due to alteration in transcription factors [erythroblast Transformation specific (Ets) and specificity protein 1 (Sp1)] or by transcriptional upregulation due to hypoxia-inducible factor activation and downregulation of repressor microRNAs (miR-1, miR-34, and miR-449a). 4 These dysregulations lead to malignant transformation (tumor growth, invasion, metastasis, and angiogenesis) through alterations in downstream cellular signaling pathways (Ras, PI3K/Akt, STAT3, and NF-κB). 5 A schematic illustration of the normal MET signaling pathway and various mechanisms underlying aberrant MET signaling pathways is represented in Figure 1.
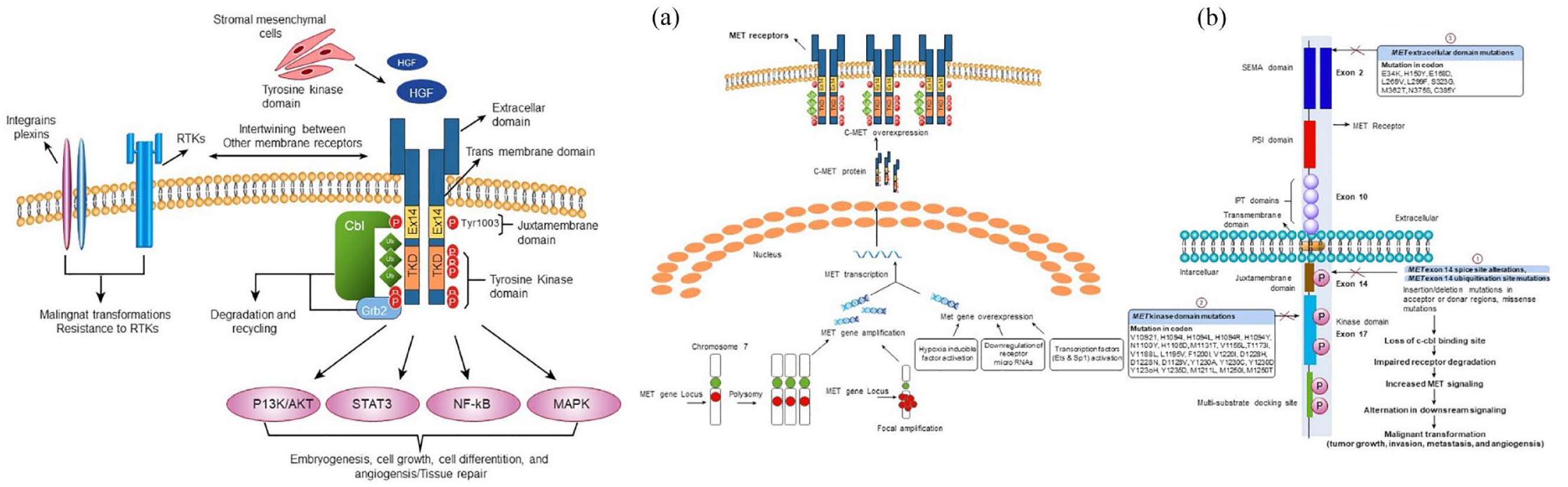
(1) Normal MET signaling pathway (left): The binding of hepatocyte growth factor (HGF) leads to receptor dimerization that activates the receptor and subsequently activates downstream signaling pathways including Ras, PI3K/Akt, STAT3, and NF-κB. This signaling basically instigates embryogenesis, cell growth, cell differentiation, angiogenesis, and tissue repair. Furthermore, downregulation of MET receptor is initiated by CBL and ubiquitin-mediated degradation, extracellular shedding as well as proteolytic cleavage. Intertwining between MET and other membrane receptors which includes plexins, integrins, EGFR, and other RTKs promotes malignant transformations and drug resistance. (2) Dysregulation of MET signaling pathway (right). Major mechanisms include (a) MET amplification (polysomy and focal amplification) and overexpression. (b) MET exon 14 skipping variants. (1) MET exon 14 splice site variants result in exon 14 exclusion, thereby lacking ubiquitin-binding site in the juxtamembrane domain and ultimately impairing MET degradation and increasing MET signaling. Furthermore, Missense mutations in the juxtamembrane domain prevent spliceosome binding and modify the Y1003 ubiquitylation site in the MET protein. (2) Mutations in the kinase domain lead to increased activation of the MET kinase and can be associated with conformational changes that favor the inactive conformation state. (3) The implications of extracellular mutations that include HGF-binding site (SEMA domain) are currently unclear. All these induce alterations in downstream signaling and ultimately induce mutagenic transformations.
In view of this pivotal role of MET in tissue remodeling and morphogenesis, scholars in several studies have reported that MET alterations, particularly MET exon 14 skipping variants, MET amplification, MET overexpression, and MET fusion, play a key role in pathogenesis and alteration in sensitivity to targeted therapies and contribute to the development of acquired tumor cell resistance to treatment with EGFR-targeting TKIs in different cancer types. 6 A few anti-MET-TKIs have been developed for MET-directed targeted therapies, such as tepotinib and capmatinib, which have been approved in the United States and Japan, respectively, and savolitinib, which was approved by the National Medical Products Administration of China in June 2021,7–9 as well as glumetinib, which has been approved in China in March 2023. In view of the importance of MET gene alterations in cancer pathogenesis and therapy, the detection of MET abnormities has become increasingly important in clinical practice to guide patient selection for targeted therapy, and testing for MET exon 14 skipping variants and amplifications has already been recommended by the NCCN guidelines in treatment-naive and TKI-resistant non-small-cell lung cancer (NSCLC) patients, respectively. 10 This review will summarize the present situation and future of MET alteration detection technology, especially exon 14 skipping, amplification testing, and overexpression testing, to provide a landscape of MET alteration-associated detection and targeted therapy updates.
MET variation
MET exon 14 skipping variants in cancers
A diverse range of variations involving the kinase domain, exon 14, intronic splice site, and SEMA domain can occur within MET (Figure 1). Furthermore, a splicing variant in MET leading to loss of MET exon 14 emerged as a biomarker and offers a potential therapeutic target in several cancer types. 11 Therefore, robust approaches for the detection of such skipping events in MET exon 14 are critical in the clinical management of cancers, specifically NSCLCs, and other cancers harboring MET exon 14 skipping variants. 3 The prevalence of MET exon 14-skipping variants is widely reported in lung cancer, with a frequency of 0.9–4%. 12 Among all cancers, the prevalence of MET exon 14 skipping variants is widely reported in NSCLCs, and the most widely used platform for the detection of this variant includes next-generation sequencing (NGS) followed by reverse transcription polymerase chain reaction (RT-PCR). A summarization of prevalence and prognosis is presented in Table 1.
Summarization on prevalence and prognosis of MET exon14 skipping variants.
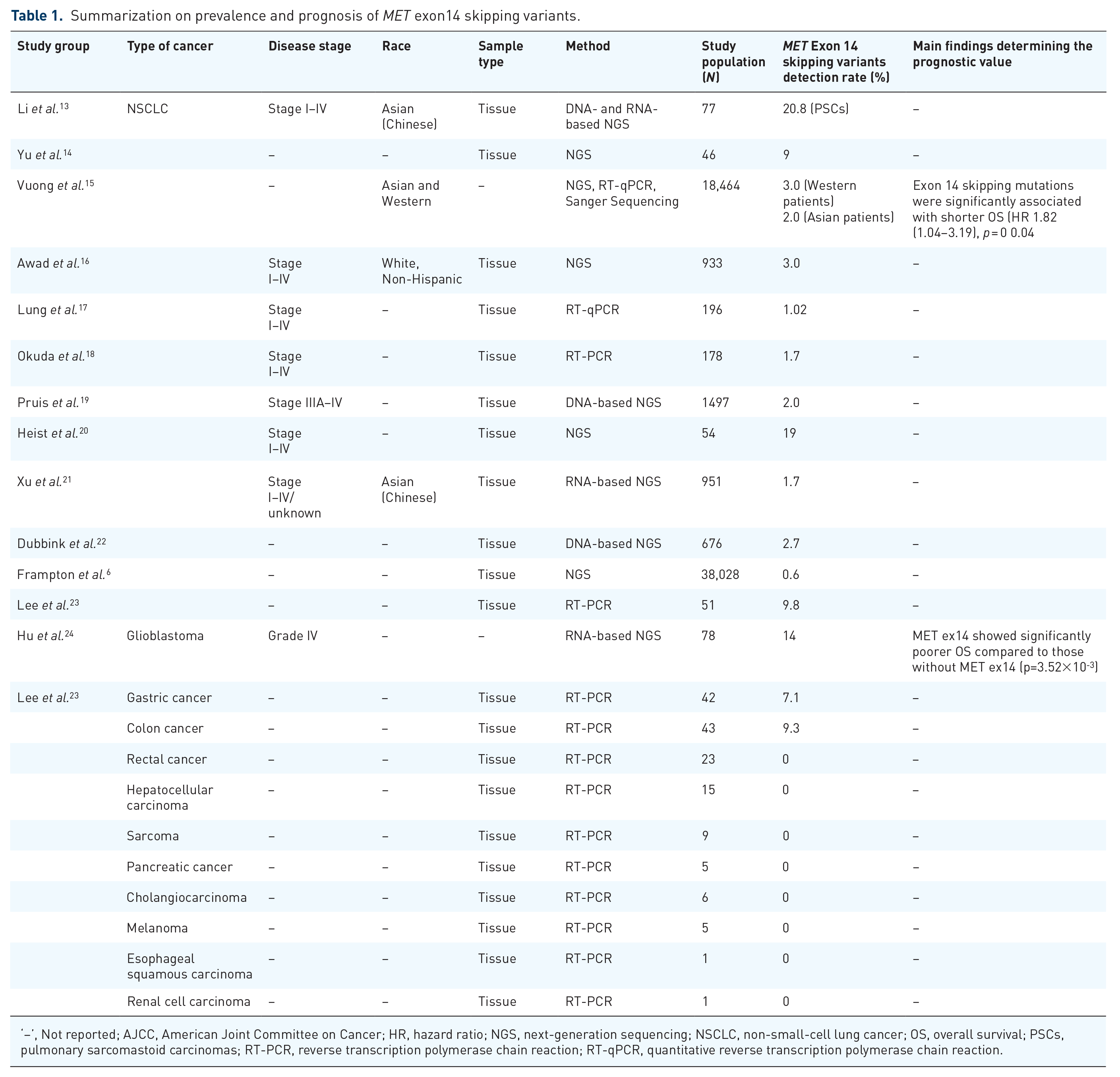
‘–’, Not reported; AJCC, American Joint Committee on Cancer; HR, hazard ratio; NGS, next-generation sequencing; NSCLC, non-small-cell lung cancer; OS, overall survival; PSCs, pulmonary sarcomastoid carcinomas; RT-PCR, reverse transcription polymerase chain reaction; RT-qPCR, quantitative reverse transcription polymerase chain reaction.
Detection of MET exon 14 skipping variants
Genome-wide sequencing revealed heterogenic forms of MET exon 14 variants at the DNA level, thereby making it challenging to detect either by amplification refractory mutation system (ARMS)-PCR or by DNA NGS panels; instead, RNA-based testing can further improve testing accuracy. 16 Furthermore, limited reports are available on the comparison of these detection platforms.
DNA-based NGS
A detailed methodology of NGS is represented in Figure 2(c). Generally, two types of NGS-based assays, namely, amplicon-based and hybrid capture-based NGS platforms differing in DNA enrichment methods, are used in clinical settings. 25 The major limitation associated with routinely used commercially available amplicon-based NGS panels for detection of MET exon 14 skipping variants includes the frequent emergence of new regions to be covered that lead to allelic dropouts and sequencing errors. 26 Earlier, Davies et al. compared amplicon-mediated DNA-based NGS versus RNA-based NGS in NSCLC tumor samples for MET exon 14 skipping variants and reported that among 286 samples tested by both assays, RNA-based testing detected 10 positive samples, 6 of which were not detected by the DNA-based assay. Further examination revealed that genomic deletion involving primer binding sequences was the likely cause of false negatives reported and led to the further conclusion that amplicon DNA-based NGS misses the detection of a substantial fraction of MET exon 14 alterations as they are located outside the amplified regions. 27 However, amplicon-based NGS has the advantage of improved capture of targets and sequencing of difficult regions with shorter turnaround time when compared to hybrid-based NGS. 3 The pitfalls associated with amplicon-based NGS can be addressed with the adoption of hybrid-based NGS, where it not only allows the identification of hotspot mutations but also interrogates entire coding sequences of oncogenes, tumor suppressor genes, and introns of selected genes that are involved in gene fusions and further allows the assessment of copy number alterations. 28 Furthermore, if the designed algorithm and probe/bait sufficiently cover the region of interest, DNA-based assays using hybrid capture-mediated target enrichment are less likely to produce false-negative results. This is exemplified by the studies from Frampton et al. and Awad et al., where a wide variety of MET exon 14 skipping variants, including large deletions, were successfully detected by employing hybrid-based NGS assays.6,16 Furthermore, a hybrid-based approach enables corrections for some of the sequencing bias and allele dropout issues associated with amplicon-based NGS assay. 29 Although the depth of coverage of genes of interest in both platforms was high, hybrid-based NGS, with its noteworthy advantage, outperforms amplicon-based NGS. 3

Schematic illustrations of workflow of various MET aberration detection techniques: (a) polymerase chain reaction, quantitative reverse transcription polymerase chain reaction (RT-qPCR) is based on conventional PCR which utilizes a fluorescent readout to measure the amount of PCR product after each round of amplification 30 ; (b) fluorescence in situ hybridization (FISH) assay involves three steps, that is, sample fixation and denaturing the sample that involves conversion of double-stranded DNA into single-stranded DNA and subsequent hybridization where denatured single-stranded DNA was tagged with fluorescent-labeled single-stranded DNA probes and followed by visualization of the hybridized probe-target DNA complexes under fluorescent microscope. 31 (c) Next-generation sequencing (NGS) is a high-throughput technology that utilizes parallel sequencing of multiple fragments to determine the sequence. It is a complicated process with multiple steps and high requirements of quality control, different NGS platforms adopt their own specific protocol in the sequencing methods. An overview of NGS workflow (both DNA based and RNA based) was represented. (d) Immunohistochemistry: It involves the utilization of anti-MET antibodies [monoclonal antibodies (SP44, cMET, and MET4) or polyclonal antibodies (MET AF276)].
RNA-based NGS or RT-PCR
MET exon 14 variants can also be detected at the RNA level. Li et al. conducted a study that involved the comparison of DNA- and RNA-based NGS for the detection of MET exon 14 skipping variants in pulmonary sarcomatoid carcinomas (PSCs) and reported a concordance of 96.1% between these platforms and concluded that RNA-based sequencing was the most accurate because some somatic variants not covering MET exon 14 splice sites might also induce skipping. 13 Furthermore, DNA sequencing cannot confirm the absence of the exon, as modifications such as splicing occur post-translationally. 27 At this juncture, RNA-based NGS platforms have the potential to complement DNA-based NGS platforms where RNA sequencing permits the direct recognition of the loss of exon 14 transcription. 27 In a study, Jurkiewicz et al. compared the performance of DNA versus RNA-based NGS assays for the detection of MET-ex14 skipping variants in 644 lung adenocarcinoma samples and concluded that DNA-based NGS panels can potentially miss MET-ex14 skipping when the primers do not target both the 3′ and 5′ splice sites of introns 13 and 14, respectively. 32 Furthermore, due to the diverse nature of MET exon 14 splice site alterations, interpreting variants that truly result in exon 14 skipping is challenging, and this becomes more strenuous when these alterations are located in deeper introns; since RNA-based platform involves sequencing mRNA that is devoid of introns, this kind of challenge can be avoided. 33
RNA-based RT-PCR can also be used for MET-ex14 skipping detection and shows good concordance with RNA-based NGS. 12 Although NGS is rapid in comparison to traditional Sanger sequencing, it is still too expensive to be affordable by small laboratories or an individual. 34 By contrast, RT-PCR is easy to perform, more widespread, and has a shorter turnaround time. RT-qPCR is usually designed in the MET exon 13 and 15 region primers for the detection of specific amplification products. This method has a high accuracy in detecting MET 14 variants but it can miss some special and rare forms of MET variations. Meanwhile, RNA-based analysis is highly reliant on the quality of RNA, which can be suboptimal in some clinical samples. 27 RNA-based testing is not part of the routine workflow in many molecular detection laboratories, as acquiring sufficient RNA material remains a large obstacle in contrast to DNA acquisition. Therefore, when RNA quality is at risk, an alternative is DNA-based NGS, where DNA is less difficult to obtain and less vulnerable to degradation.
Targeted therapies for MET exon 14 skipping variants
The constant discovery of actionable activating variants has led to targeted therapies with new-generation TKIs and improved overall survival and time to progress. 26 Among all reviewed literature, NSCLC lung cancer accounts for almost all. Earlier, a phase I PROFILE 1001 study reported the efficacy of crizotinib [median progression-free survival (mPFS) 7.3 months and overall response rate (ORR) of 32%] in advanced-stage NSCLC patients harboring MET exon 14 alterations, and these results led to the inclusion of crizotinib in the NCCN guidelines. With this breakthrough, several highly selective MET inhibitors, such as tepotinib, capmatinib, savolitinib, and glumetinib, have been tested in patients with MET exon 14-altered NSCLC and found to be more potent than crizotinib. 35
Tepotinib was developed for the treatment of solid tumors and demonstrated promising clinical efficacy and safety profiles in patients with advanced NSCLC with a confirmed MET exon 14 skipping variant in the multinational phase II VISION study. 36 In March 2020, it was approved for use in Japan for this indication and was subsequently approved by the FDA on 3 February 2021. Capmatinib was developed for the treatment of lung cancer, inhibiting cancer cell growth driven by the mutant MET variant, including exon 14 skipping. In May 2020, capmatinib received its first global approval for the treatment of adults with metastatic NSCLC with MET exon 14-skipping variants as detected by an FDA-approved test. 8 Savolitinib was developed for the treatment of NSCLC, gastric cancer, colorectal cancer, and papillary and clear cell renal cell carcinoma. Based on the results of a pivotal phase II trial, savolitinib yielded promising activity and had an acceptable safety profile in patients with NSCLC/pulmonary sarcomatoid carcinoma and was recently approved in June 2021 in China for the treatment of metastatic NSCLC with MET exon 14-skipping alterations in patients who have progressed after or who are unable to tolerate platinum-based chemotherapy, conditional on the results of a phase III trial. 37 Glumetinib was developed for the treatment of lung cancer and showed durable antitumor activity with manageable toxicity in patients with locally advanced or metastatic MET ex14-positive NSCLC in the phase II GLORY study. 38 In March 2023, it was approved in China for this indication.
Furthermore, reports from several clinical studies cannot be compared directly due to different populations and inclusion criteria in the clinical efficacy of several MET-TKI inhibitors, including tepotinib 39 [independent review committee (IRC): ORR 57.3% (treatment naive), 45.0% (pretreated), 40 capmatinib (ORR: 44% (pretreated) and ORR: 68.3% (treatment naive) 35 , mPFS 12.5 m (treatment naive), 5.5 m (pretreated); mOS 25.5 m (treatment naive), not reported (pretreated))], 41 and savolitinib [phase IIIb study, IRC: ORR 58.6% (treatment naive), mPFS 13.8 m (treatment naive) 42 ; phase II study(36% PSC and 21% CNS metastases): TRES set: ORR 49.2%, FAS set: mPFS 6.9 m, mOS 12.5 m; in PSC, mPFS 5.5 m, mOS 10.6 m; other NSCLC (non-PSC), mPFS 7.0 m, mOS 17.3m 43 ] and glumetinib 38 (BIRC: overall ORR 66%, mPFS 8.5 m, mOS 17.3 m; in treatment-naïve patients: ORR 71%, mPFS 11.7 m) in patients harboring MET exon 14 skipping variants in NSCLC. A summarization of MET exon 14 skipping variant targeted therapy outcomes in advanced NSCLC is represented in Table 2.
MET exon 14 skipping variants targeted therapy outcomes in advanced NSCLC.

mPFS, median progression free survival; NGS, next-generation sequencing; NR, not reached; NSCLC, non-small-cell lung cancer; ORR, overall response rate; PFS, progression-free survival; PSCs, pulmonary sarcomatoid carcinomas; RT-PCR, reverse transcription polymerase chain reaction.
MET amplification
MET amplification in cancers
MET GCN gain occurs either by polysomy or focal amplification (Figure 1).3,47 Tumors harboring de novo MET amplifications (high level) are primarily dependent on the MET signaling pathway for growth and are found across a wide variety of solid tumors, 3 while tumors harboring acquired MET amplifications rely on other oncogene alterations (such as EGFR mutation) and develop a secondary dependence on the MET signaling pathway as a mechanism of resistance to targeted EGFR therapies. 48 Reports from several studies have indicated that MET gene amplification drives treatment resistance, particularly therapies targeting EGFR.49–53
MET amplification occurs in a broad type of cancer as protooncogenes or resistance mechanisms. A summarization of the prevalence and prognosis of MET amplification in various cancers is presented in Table 3.
Summarization on prevalence and prognosis of MET amplification in various cancers.

‘–’, Not reported; AJCC, American joint committee on cancer; cfDNA, circulating cell-free DNA; GC, gastric carcinoma; HR, hazard ratio; mCRC, metastatic colorectal cancer; NSCLC, non-small-cell lung cancer; PRCC, papillary renal cell carcinoma; RT-PCR, reverse transcription polymerase chain reaction; RT-qPCR, reverse transcription polymerase chain reaction; FISH, fluorescence in situ hybridization.
Regarding polysomy, there is controversial evidence regarding whether MET polysomy is the driver of gene alteration for tumorigenesis. Lai et al. found that although up to 26% of TKI-naive EGFR mutant-positive NSCLC harbor high MET GCN by FISH, this did not significantly affect response to TKIs, except in patients identified as MET-amplified but not polysomy. 74 In the phase II Acse study, advanced NSCLC patients with MET polysomy and low amplification (MET/CET7 ratio between 1.8 and 2.2) did not respond well to crizotinib, with a mPFS of 3.2 months and a mOS of 7.7 months. 45 However, in the phase 1b/2 TATTON study, osimertinib and savolitinib showed encouraging antitumor activity in both MET amplification and polysomy patients recruited based on FISH positivity, with an ORR of 30% in part B1 (after prior 3G TKIs). 12 The subgroup analysis results showed that the response of focal amplification was better than that of polysomy (ORR 31% versus 28%) but more research is needed for verification. Therefore, MET amplification may be considered as a biomarker for MET-directed therapies, while polysomy still needs to be studied more.
Detection of MET amplification
MET amplification (copy number gain) can be detected by several techniques, such as FISH, NGS, and ddPCR. 3 As the magnitude of MET amplification is a continuous variable, determining the cutoff for MET positivity is more challenging. 3 At present, no consensus exists on the most appropriate diagnostic cutoff point for MET amplification. 75 Different detection platforms for MET amplification testing exist and have ineligible disparities in terms of sensitivity and specificity.
Fluorescence in situ hybridization (FISH)
FISH is a cytogenetic technique used for obtaining spatial genomic and quantification of nucleic acids in the cellular environment and has emerged as the gold standard technique for the detection of chromosomal abnormalities. 76
In the FISH assay, MET copy number increases can be defined either by Cappuzzo criteria or by University of Colorado Cancer Center (UCCC) criteria, which calculate the ratio of MET to chromosome enumerating probe against chromosome 7 (CEP7). The Cappuzzo et al. criteria define MET amplification as a mean of five or more copies of MET per cell (MET gene copy number (GCN) ⩾5). 77 Furthermore, other alternate definitions suggest a MET GCN of ⩾6 78 and a MET GCN of ⩾15. 79 However, the determination of GCN cannot differentiate between MET focal amplification and polysomy, and this limitation is overcome by the UCCC approach involving the calculation of the MET-to-CEP7 ratio that adjusts the number of chromosomes present, thereby differentiating selective MET focal amplifications and chromosomal duplication. In general, a MET to CEP7 ratio ⩾ 2.0 defines MET focal amplification. 73 However, several other studies categorized the degree of MET focal amplification into low (⩾1.8–⩽2.2), intermediate (>2.2–<5), and high (⩾5). 75
The FISH technique is advantageous in the detection of MET amplification in light of its comparable performance characteristics and potential for cost-effectiveness and limited complexity in testing when compared with NGS. Furthermore, FISH offers direct visualization of the tested samples, which was not possible by NGS, and another added benefit of this technique is that it represents a suitable technology for the detection of intratumoral heterogeneity within tissue samples. Although FISH is the current gold standard for MET amplification testing, the prevalence of MET amplification detected by FISH is variable in the literature, which is likely attributable to a lack of standardization of technique and/or patient selection criteria and different cutoffs for defining MET positivity. 63 In addition, the main limitation of FISH is that it can only be applied to tissue samples, while tissue feasibility is low in advanced NSCLC patients, especially patients who progressed on previous TKIs.
Next-generation sequencing
Similar to the FISH assay, there is no consensus on a single definition of MET amplification, and the cutoff for MET amplification varies across different NGS platforms. 80 Hybrid capture-based NGS is known to be more accurate in assessing copy number variation in MET and other genes, as it interrogates broader regions of the genome and removes sequence replicates. By contrast, amplicon-based NGS covers a limited genomic territory, and sequence replicates cannot be removed, affecting sequence coverage depth. 3 NGS can be used to analyze both tissue and plasma ctDNA or other bodily fluids, which will facilitate biomarker testing extensively. However, certain technical limitations, including sample selection, low tumor cell fraction, and low DNA quality of tumor samples can increase background noise, hindering the accurate analysis of copy number gain/loss. 80 As reported in the TATTON study, where osimertinib (3rd EGFR-TKI) and savolitinib (MET-TKI) combined therapy in NSCLC patients with MET-amplified EGFR-TKI resistance demonstrated encouraging antitumor activity, FISH positivity was defined centrally as either focal amplification (MET:CEP7 ratio ⩾ 2) or polysomy (gene copy number ⩾ 5 if MET:CEP7 <2), while tissue NGS from Foundation Medicine showed higher positive-percent agreement (PPA, also known as sensitivity) for FISH focal amplification (88%) but lower PPA for FISH polysomy (4%). Similarly, a comparison of ctDNA NGS with FISH yielded negative-percent agreement (NPA) of 90% and a modest PPA of only 25%. 45 Various factors affect the sensitivity of ctDNA detection by NGS, such as sequencing depth, threshold of bioinformatic analysis, and techniques to enrich tumor-derived signals and reduce background noise. The high specificity and low sensitivity of the currently available NGS assay indicate that copy number variation (CNV) detection by NGS needs to be optimized, and retesting with FISH should be considered to avoid missing MET amplification; while NGS may serve as an alternative selection for MET polysomy with higher sensitivity and specificity and guide the clinical treatment. 81
Although plasma ctDNA testing of MET amplification is challenging, it is a direction for future development to fulfill the clinical needs given that plasma specimens are dominant in late-phase patients. In addition, plasma harvested from peripheral blood is less invasive and can reflect the disease progression dynamically while avoiding tumor heterogenicity in single surgery or biopsy samples. 82
Polymerase chain reaction
MET amplifications can be detected by employing a PCR technique specifically (RT-PCR). Similar to FISH and NGS, the cutoff for defining MET amplification positivity was not standardized. 62 A comparison of the basic principle and procedure involved in different PCR techniques is represented in Figure 2(a). The major limitation associated with RT-PCR is that the success relies on the RNA quality in the specimen tested. Inadequate fixation or prolonged ischemia of tissue leads to false-negative results. These limitations were outweighed by the introduction of droplet digital PCR (ddPCR), which has high sensitivity and accuracy levels for absolute representation of a given nucleic acid sequence. Droplet digital PCR enables the absolute quantification of nucleic acids present in the sample by partitioning the sample into independent PCR subreactions where each partition contains a few or no target sequences and is subjected to PCR wherein the fraction of amplification positive partitions is used to quantify the concentration of the target sequence with a statistically defined accuracy using Poisson’s statistics. 83
The performance of ddPCR for MET amplification testing is not well characterized compared to FISH and NGS. 84 However, the consistency between ddPCR and FISH is reported in some small sample studies for assay analytical validation of developed ddPCR methodology; the sensitivity and specificity of tissue ddPCR and FISH are both 100%, while the sensitivity and specificity of tissue and blood ddPCR are 66.67% and 98.86%, respectively. 85 Given that insufficient tissue can be retrieved after resistance to EGFR-TKIs, further studies to confirm the testing capability of blood ddPCR as an alternative detection tool for MET amplification is needed in the future.
Targeted therapies for MET amplification
The prevalence of acquired MET amplification is enriched gradually after lines of EGFR-TKI treatment. In lung cancer, MET amplification occurred in 1−4% of treatment-naive patients, while the prevalence increased to 5−22% after first- and second-generation TKI treatment and 5−50% after third-generation TKI osimertinib treatment. 86 The amplification of MET occurred independently of the EGFR T790M mutation and was clinically relevant to gefitinib and erlotinib resistance. 60
There are several MET-TKIs in development for MET amplification-positive NSCLC patients, such as crizotinib, tepotinib, capmatinib, and savolitinib, and criteria for patient inclusion are mainly based on FISH and immunohistochemistry (IHC). The cutoff for MET amplification positivity varies among studies 83 Furthermore, it is evident from the literature that patients harboring higher MET GCN achieve better clinical outcomes from the targeted therapy.
For de novo MET amplification NSCLC, in a phase I PROFILE 1001 study in which NSCLC patients were stratified based on the degree of MET amplification and the activity of crizotinib examined in relation to the level of MET amplification, there was a high amplification group (MET-to-CEP7 ratio ⩾ 4) with reported ORR of 38.1% and median PFS of 6.7 months compared with a low amplification group (MET-to-CEP7 ratio ⩾ 1.8 to ⩽2.2) with ORR of 33.3% and median PFS of 1.8 months and an intermediate amplification group (MET-to-CEP7 ratio > 2.2 to <4) with ORR of 14.3% and median PFS of 1.9 months. 87 Earlier, the results of the GEOMETRY mono-1 study demonstrated the efficacy and safety of capmatinib in patients with high-level (GCN) ⩾10 compared with low-(GCN < 4) or mid-level (GCN 4–5 or 6–9) MET-amplified advanced NSCLC. Patients with GCN ⩾ 10 treatment-naive and/or receiving 1 or 2 lines of therapy exhibited better ORRs of 40% and 29%, respectively. 35 It seems that high amplification status is associated with better response to MET-TKIs compared to low amplification status given the current evidence. Recently, the results from the VIKTORY umbrella trial showed that savolitinib monotherapy exhibited an ORR of 50% (10/20) in a subset of gastric cancer patients harboring MET amplifications, and further genomic analysis revealed that patients with MET GCN > 10 (by tissue NGS) had high response rates to savolitinib [ORR 70% (7/10)] and concluded that the subset of patients with MET amplifications achieved the largest absolute decrease in tumor burden. 83 Furthermore, despite there existing evidence of MET inhibition by foretinib, Shah et al. reported disappointing results for foretinib, which might be due to disparities in the selection of the study population, study design, or drug itself. 88 Furthermore, a summarization of MET amplification targeted therapy outcomes by MET copy number status across different cancer types is represented in Table 4.
MET amplification targeted therapy outcomes.

‘–’, not reported; CEP 7, chromosome 7; FISH, fluorescence in situ hybridization; GCN, gene copy number; IHC, immunohistochemistry; NSCLC, non-small-cell lung cancer; PRCC, papillary renal cell carcinoma; NGS, next-generation sequencing; PFS, progression-free survival; ORR, overall response rate.
As shown in Table 4, the commonly used platform for MET amplification testing and patient screening is FISH, combined with the IHC platform for some studies. The criteria for MET amplification varied across studies, and a unified cutoff has not yet been established. Both the MET-to-CEP7 ratio and MET GCN number were used separately or combined. It seems that a better response to MET-TKIs or combined therapy occurs in patients with higher MET amplification status.
MET overexpression
MET overexpression in cancers
MET overexpression can be caused by gene amplification, gene mutation, transcriptional enhancement [activation of specificity protein 1 (Sp1), erythroblast transformation specific (Ets)], or by post-transcriptional mechanisms that lead to malignant transformations (Figure 1). 4
The clinicopathological impacts of MET overexpression in various cancers have been investigated in several studies. The prevalence of MET overexpression was reported to be 39.8%, 100 33.7%, 101 and 58.8% 102 in cases of gallbladder carcinoma, triple-negative breast cancer, and NSCLC, respectively. However, the potential correlation of MET overexpression with patient outcome is inconsistent across tumors. Some of the studies indicated that MET overexpression was significantly related to shorter OS or PFS in bladder cancer 103 and glioblastoma multiforme, 104 while some indicated no correlation to prognosis in NSCLC 102 or lung adenocarcinoma 105 Thus, a detailed understanding of the relationship between MET overexpression and prognosis is still needed.
Detection of MET overexpression
MET overexpression can be analyzed using immunohistochemistry (IHC), which provides a semiquantitative information on MET expression. 3 The prevalence rate of MET overexpression is approximately 13.7−63.7% in all NSCLCs. Among them, the prevalence rate of MET overexpression is approximately 30.4−37% in advanced NSCLC after EGFR-TKI treatment. Different scoring systems were used to quantify the level of MET expression by IHC. 106 In clinical trial settings, the level of expression is quantified as a clinical score (on a scale of 0–3+). The H-score (range from 0–300) is another scoring system that involves multiplying the percent of cells with staining scores of 1+, 2+, and 3+ by their staining intensity score. 107 In general, an H-score ⩾ 200 denotes overexpression, and the specific cutoff range varies among studies. 108 Recently, a Chinese expert consensus on MET immunohistochemistry detection and interpretation standards for NSCLC has been proposed, aiming to improve the quality of detecion and interpretation to further guide the clinical treatment and studies. 109
Some MET IHC antibodies for MET overexpression testing are shown in Table 5, including SP44, D1C1, and 3077. At present, many domestic and foreign antibodies for detecting MET amplification have obtained domestic medical device product status (recorded in the National Medical Products Administration), involving multiple clone numbers. The staining performance of different antibodies varies, and the interpretation criteria of current clinical research combine the expression intensity and percentage of relevant antibodies in tumor cells at the same time.
MET overexpression targeted therapy outcomes.
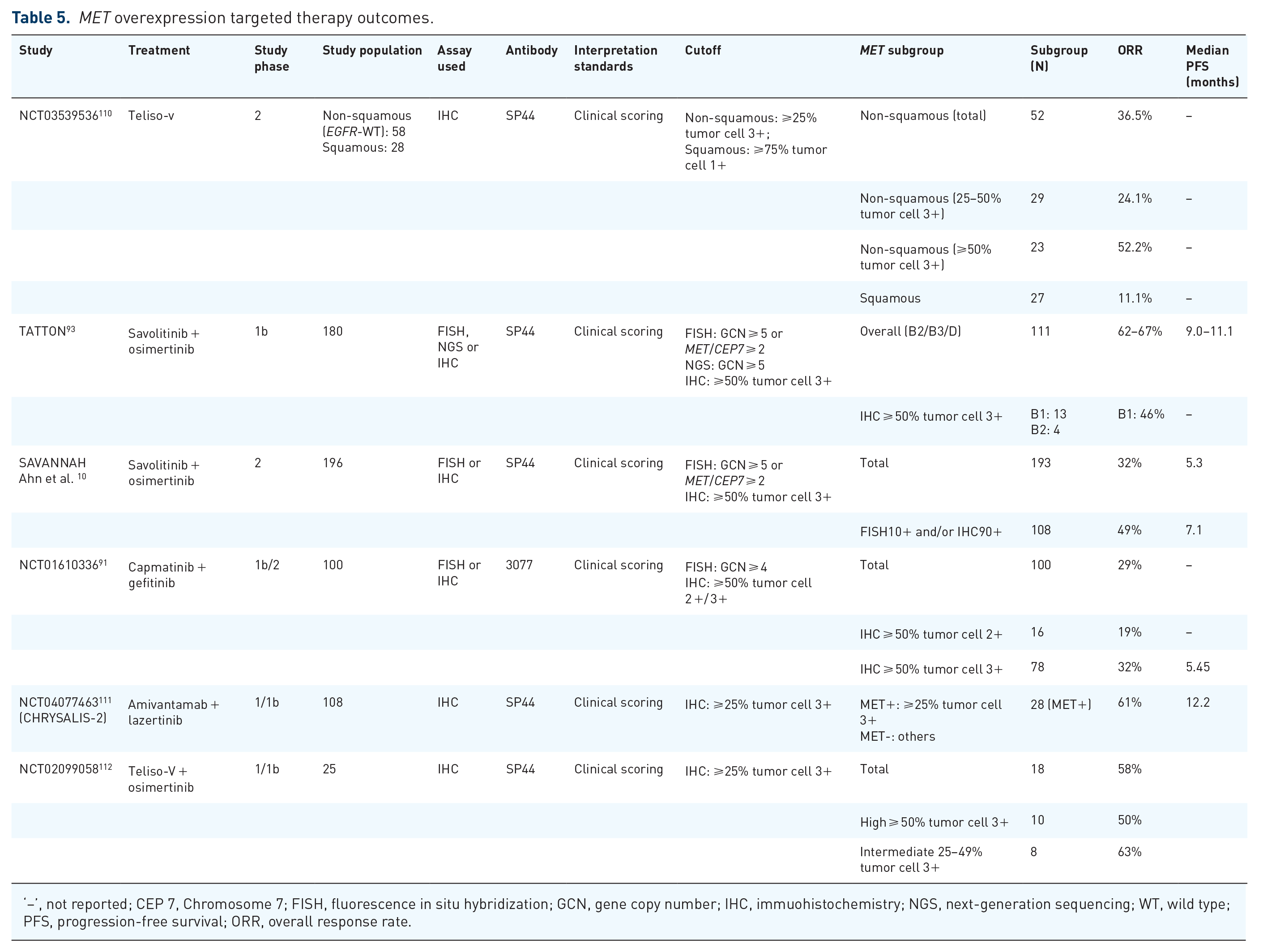
‘–’, not reported; CEP 7, Chromosome 7; FISH, fluorescence in situ hybridization; GCN, gene copy number; IHC, immuohistochemistry; NGS, next-generation sequencing; WT, wild type; PFS, progression-free survival; ORR, overall response rate.
Targeted therapies for MET overexpression
MET can be overexpressed in certain cancers harboring primary and/or secondary MET exon 14 alterations or MET amplifications, 3 and many studies have indicated that MET overexpression and gene amplification are prognostic survival factors for many cancers, including gastric carcinomas. 113 At this juncture, the results from several clinical trials involving monotherapy with anti-MET antibodies (onartuzumab, emibetuzumab), anti-HGF antibodies (ficlatuzumab, rilotumumab), TKIs (crizotinib, tivantinib, cabozantinib), and other therapeutic agents suggest that the overall activity of these monotherapies in MET-overexpressing cancers is low, indicating that MET overexpression is not consistently predictive of benefit from MET-directed therapies. 114
By contrast, combination with EGFR-directed therapies is effective. Wu et al. conducted an INSIGHT trial aiming to evaluate the efficacy and safety of tepotinib plus gefitinib in NSCLC patients harboring MET overexpression or MET amplification and acquired resistance to previous EGFR inhibitors and concluded that patients with MET IHC3+ or MET amplifications showed a better response, where PFS and OS were longer with tepotinib plus gefitinib (PFS 8.3 months, OS 29.1 months) than with chemotherapy (PFS 4.4 months, OS 17.9 months) in patients with IHC3+ (⩾50% tumor cells with strong intensity) MET overexpression, while the IHC2+ (⩾50% tumor cells with moderate intensity) subgroup showed a poor response to combination therapy with a MET inhibitor and EGFR inhibitor. 115 Overall, IHC is the only detection method for MET expression in clinical trials. The cutoff for MET expression varies across studies, and 50% of tumor cells 2+/3+ are usually used as criteria. Among the studies in Table 5, subgroup analysis suggested that a higher percentage of tumor cell 3+ or the same percentage with a higher staining score generally improved clinical outcomes. Therefore, given the limited clinical studies and a small number of patients, higher MET overexpression can reflect better outcomes for MET-TKIs plus EGFR-TKIs treatment in EGFR-resistant advanced NSCLC patients harboring MET overexpression. More studies and specific diagnostic criteria for MET overexpression are required to identify patients who can benefit more from combination therapy with MET-TKIs and EGFR-TKIs in this setting.
Furthermore, MET overexpression is not a reliable indicator of MET exon 14 alterations or MET amplifications, and reports from several studies have indicated the same where MET overexpression determined by IHC does not correlate with MET amplification. 116 This difference in correlation might be due to the inclusion of samples (featuring a low level of amplification) that do not result in considerable protein expression or protein expression modulation by posttranscriptional and posttranslational factors. 3 Therefore, patients harboring activating alterations in MET can be investigated for the presence of MET overexpression, but unfortunately, MET overexpression is not a reliable indicator of MET amplifications/MET exon 14 skipping variants. 3 Furthermore, a growing number of clinical trials are in the pipeline to explore the relationship between MET expression and MET amplification; however, unifying guidelines for standard scoring systems for IHC are required to obtain consensus among different trials.
MET fusion
MET fusion in cancers
The MET fusion was first found in chemically transformed osteosarcoma cell lines, which was the TPR-MET fusion. 3 Thereafter, MET fusions were identified in a variety of tumors over the years, such as gastric cancers, lung adenocarcinoma, thyroid carcinoma, hepatocellular carcinoma, and glioma. 3 Beyond TPR-MET, multiple other fusions have been identified, including PTPRZ1-MET, CLIP2-MET CAPZA2-MET, ST7-MET, TRIM24-MET, KIF5B-MET, RBPMS-MET, and EML4-MET, most of which have been reported in case reports.117–122 The exact frequency of MET fusion in these cancers is poorly defined; of them, glioma had the highest proportion at 15%. 123 As a result, MET fusions and their therapeutic implications have been largely ignored. MET fusions have rarely been described in NSCLC, with an overall frequency of approximately 0.29%, and half of the fusion types are intragenic fusions.122,124 A large real-world multicenter study for the Chinese population detected putative MET fusions with a prevalence of 0.15% in 79,803 solid tumors, while the majority of them were lung cancer patients (75.4%). 125 It is worth considering that some patients with MET fusion can benefit from MET-TKI therapy.
Detection of MET fusion
Numerous assays can detect MET fusions, including FISH, RT-PCR, and NGS. However, complex/novel rearrangements may result in inadequate FISH for detecting many MET fusions. 3 Therefore, NGS has become the increasingly preferred assay in the clinic.
NGS with traditional amplicon-based enrichment is less accurate in identifying gene fusions with unknown partners, while a technique termed anchored multiplex PCR (AMP) possibly addresses this limitation. 126 In AMP, a ‘half-functional’ NGS adapter is ligated to cDNA fragments that are derived from input RNA, and then the amplification between gene-specific primers and a primer to the adapter leads to target enrichment. As a result, the gene fusions of interest, even if they involve a novel fusion partner, should be detected. 126 Currently, targeted RNA-based NGS (tRNA-seq) is increasingly being applied in molecular detection for gene fusion in solid tumors, which is efficient for the simultaneous detection of actionable gene fusions, splice variants, single nucleotide variants (SNVs), and indels. 127 RNA-seq is not only a well-validated tool for detecting gene fusions in fresh-frozen tumors but also in formalin-fixed, paraffin-embedded (FFPE) tumor samples. It showed a sensitivity of 83.3% in clinical FFPE specimens, with a negative prediction value of 94.3%, and was regarded as a complement DNA-based NGS assay. 128
In NSCLC, sequentially combining DNA NGS and RNA NGS was shown to be one of the most efficient strategies for fusion detection; it was feasible on small tissue samples and could drastically reduce the complexity and cost of molecular workup. 129 Song et al. 130 developed a convenient single-tube, dual-template assay, and an integrated bioinformatics pipeline for relevant variant calling, in which RNA was used for fusion detection, whereas DNA was used for SNVs and insertion and deletions (indels). This method was considered to benefit not only most patients carrying target fusion but also those with rare variations. 130 Wei et al. 131 designed a lung-cancer-specific targeted all-in-one transcriptome-based assay based on single primed enrichment technology which covered gene loci that are related to selecting optimal targeted therapy in advanced NSCLC and could simultaneously identify mutations, gene fusions, and exon skipping events. This assay was shown to identify all the expected mutations at the transcriptome level and to reach an accuracy of close to 100%. 131
Targeted therapies for MET fusion
Precisely targeted therapy has been incredibly underexplored in MET fusion-positive cancers. Nevertheless, in recent years, there have been many clinical case reports presenting the potential for MET-TKI therapy. Among these, crizotinib (monotherapy or combination therapy) has been described as having surprising clinical responses in patients with a variety of MET fusion-positive glioblastoma and lung adenocarcinomas, including the gene fusion types CUX1-MET, HLA-DRB1-MET, CAV1-MET, ARL1-MET, PRKAR1A-MET, bringing substantial tumor shrinkage and associated relief of symptoms.132–137 Blanc-Durand et al. 138 reported a patient with NSCLC with brain metastasis harboring an HLA-DRB1-MET gene fusion who successively received crizotinib and cabozantinib and the selective inhibitor tepotinib and experienced rapid responses associated with a tremendous improvement in physical function during each treatment cycle. The potential role of capmatinib was also reported in a patient with chemotherapy-refractory metastatic cholangiocarcinoma harboring a CAPZA-2-MET fusion. 139 Kang et al. 140 attempted to explain the potential resistance mechanisms of MET inhibitors in patients with de novo MET fusions and found that secondary mutations D1228H/N or D1246N are worth further exploration. Multiple clinical trials are ongoing to evaluate the efficacy of MET-TKIs in tumor patients with MET fusions (NCT03993873, NCT02978261, NCT01639508, and CTR20181664 141 ).
Conclusion
The pivotal role of MET aberrations as a predictive biomarker of drug response has been reported in several clinical trials. Furthermore, several MET inhibitors demonstrated clinically meaningful efficacy in different cancers harboring MET alterations. Therefore, MET exon 14 skipping variant testing has gained prominence and has already been recommended in guidelines where capmatinib, tepotinib, and savolitinib have been approved for the treatment of NSCLCs. Furthermore, other small-molecule inhibitors, including cabozantinib and crizotinib, are in the pipeline. The literature suggests that assays such as NGS (DNA based and RNA based) could be a potential testing method in terms of sensitivity and operational procedures for the detection of MET alterations, specifically MET exon 14 skipping variants, in both tissue samples and plasma ctDNA, but may have limitations for CNV testing. In addition, the FISH assay remains a robust technique for MET amplification detection. However, it can be used only for single-gene tests and tissue samples, while NGS represents the future trend of testing choice in multialteration (MET exon 14 skipping variant, amplification, and fusion) multigene analysis and in situations of limitation to plasma samples. NGS seems to be a promising testing option. ddPCR is being developed for MET amplification testing, especially in blood. MET amplification and MET overexpression are continuous variables, so clinically meaningful cutoff points need to be standardized, particularly the cutoff for MET overexpression. MET overexpression is an emerging biomarker for MET-TKI treatment since an increasing amount of clinical data have been released to guide the treatment. Furthermore, prospective studies involving a wide range of cancer types and larger sample sizes are required in this direction for definite conclusions and to extend the spectrum of MET-targeted therapy.