
Abstract
Gastrointestinal stromal tumor (GIST) is the most common malignant neoplasm of mesenchymal origin. GIST spans a wide clinical spectrum that ranges from tumors with essentially no metastatic potential to malignant and life-threatening spread diseases. Gain-of-function mutations in KIT or PDGFRA receptor tyrosine kinases are the crucial drivers of most GISTs, responsible for tumor initiation and evolution throughout the entire course of the disease. The introduction of tyrosine kinase inhibitors targeting these receptors has substantially improved the outcomes in this formerly chemoresistant cancer. As of today, five agents hold regulatory approval for the treatment of GIST: imatinib, sunitinib, regorafenib, ripretinib, and avapritinib. This, in turn, represents a success for a rare neoplasm. During the past two decades, GIST has become a paradigmatic model in cancer for multidisciplinary work, given the disease-specific particularities regarding tumor biology and tumor evolution. Herein, we review currently available evidence for the management of GIST. This clinical practice guideline has been developed by a multidisciplinary expert panel (oncologist, pathologist, surgeon, molecular biologist, radiologist, and representative of patients’ advocacy groups) from the Spanish Group for Sarcoma Research, and it is conceived to provide, from a critical perspective, the standard approach for diagnosis, treatment, and follow-up.
Keywords
Gastrointestinal stromal tumor (GIST), the most frequent sarcoma subtype, 1 originates in the digestive tract and is primarily originated by gain-of-function mutations in KIT or PDGFRA receptor tyrosine kinases (RTKs).2,3
In the past two decades, GIST has become a paradigmatic model for the rational and successful development of molecularly targeted agents in cancer. As a result, five tyrosine kinase inhibitors (TKIs) hold regulatory approval for the treatment of metastatic GIST: imatinib, sunitinib, regorafenib, ripretinib, and avapritinib. 4 The benefit of these agents has resulted in a meaningful increase in overall survival (OS), from less than 12 months to more than 5 years, 5 which, in turn, constitutes a remarkable success for a rare neoplasm. Together, these advances have reshaped the clinical management of GIST patients, being currently developed in multidisciplinary teams with intertwined and complementary expertise. Given the increased complexity and the recent advances in the field, herein we review and update our prior Grupo Español de Investigación en Sarcomas/Spanish Group for Sarcoma Research (GEIS) guidelines for the management of GIST.6–8
Epidemiology and clinical presentation
The annual incidence of GIST in Spain, 1.24 cases/100,000, 9 is similar to that reported worldwide, ranging between 1.1 and 1.5/100,000. 10 GISTs occur predominantly in middle-aged and older individuals, with a peak in the seventh decade of life. There is an equal distribution across all sex, geographic, racial, and ethnic groups. The diagnosis of GIST is commonly initiated through nonspecific symptoms such as abdominal pain, gastrointestinal bleeding, or obstruction. GIST arises in the stomach (55%), small intestine (31%), colorectal (6%), esophagus (<1%), or extra-gastrointestinal organs (<5%). 10 Metastases are present at the time of diagnosis in approximately 15% of the patients and are found typically in the peritoneum and liver.
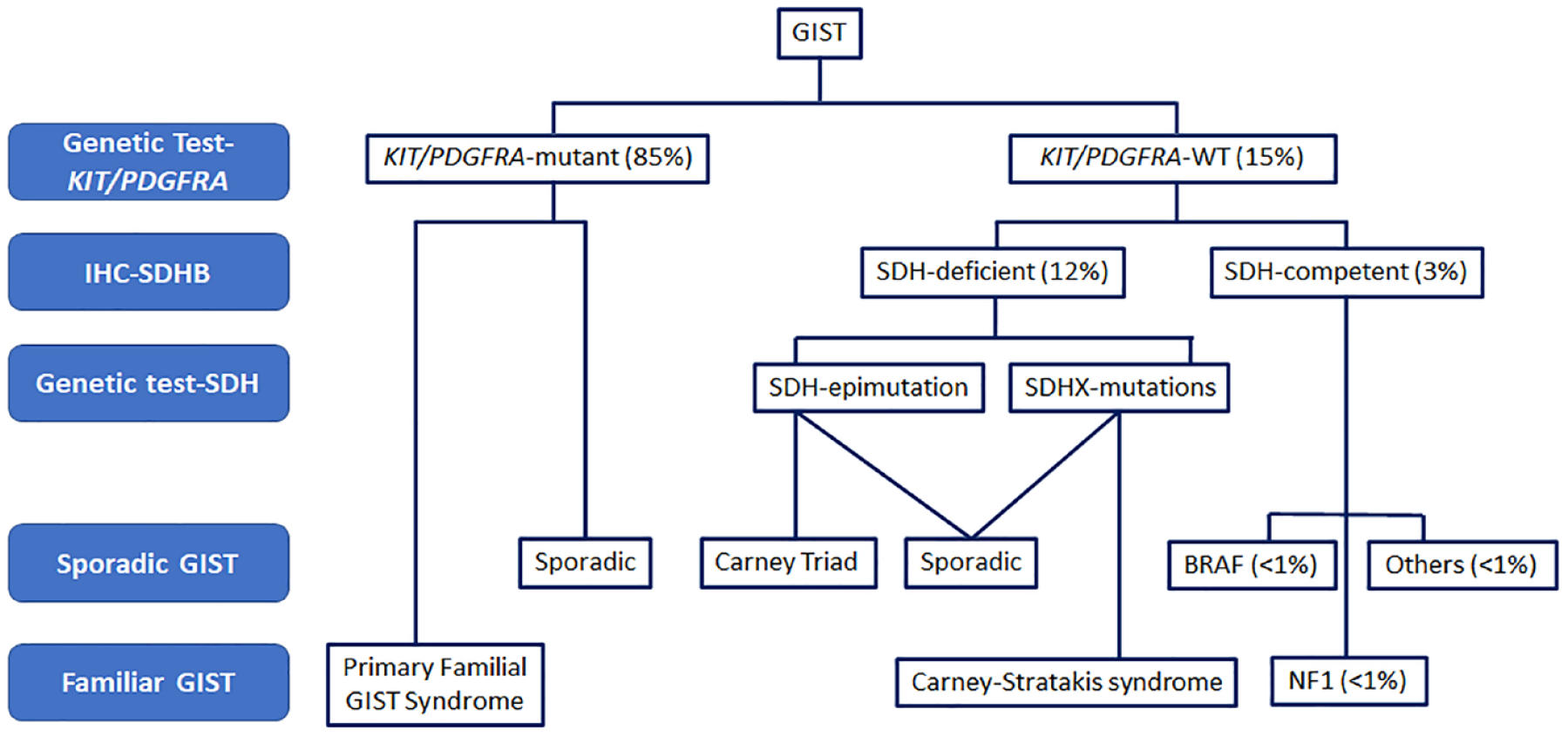
The large majority of GISTs are sporadic and lack known etiologic factors. Nonetheless, few hereditary conditions predispose to an increased likelihood of GIST (Figure 1). Carney-Stratakis syndrome is associated with germline inactivating mutations in the succinate dehydrogenase (SDH) subunits B, C, or D, leading to an early presentation of multifocal GIST, with gastric predominance, slow growth, and the possibility of developing paragangliomas. 11 In addition, 7–10% of patients with neurofibromatosis type I (NF1) develop NF1-associated GIST that typically grows as multiple implants across the small bowel. 12 Familial GISTs due to KIT or PDGFRA germline mutations are extraordinarily rare 13 and can be associated with skin hyperpigmentation, dysphagia, and mast-cell disorders. The referral of these patients to genetic counseling units is highly recommended. Finally, we and others demonstrated a 2.5- to 4-fold incidence of second neoplasms after the initial diagnosis of GIST,14,15 although there is so far insufficient evidence for these patients to be regarded as an inherited condition; therefore, it is generally not recommended to refer them, unless specific patient and family features could be relevant.
Molecular alterations in sporadic and familial GIST.
Diagnostic evaluation
Imaging
Radiological diagnosis in GIST is comparable to that performed in other digestive tract tumors. Ultrasound studies show GISTs as submucosal and hypoechogenic lesions that, if enlarged, can displace neighboring structures and turn cystic, necrotic, or hemorrhagic. An abdominal computerized tomography (CT) scan is the first choice to study location and extension. Primary tumors appear as well-circumscribed exoluminal masses that can show heterogeneous contrast enhancement representing necrotic-hemorrhagic or degenerative areas. 16 CT scans with contrast and image acquisitions of the arterial and portal phases are indicated to identify the subcentimetric hypervascular hepatic lesions that would otherwise be initially unnoticed – but become hypodense if responding with first-line TKI therapy. Magnetic resonance imaging is indicated in the assessment of rectal GIST, while positron emission tomography (PET)-CT is rarely used during the initial workup. 17
Biopsy
A preoperative biopsy is not necessary when the assessment of the primary lesion is considered suspicious of GIST and deemed surgically resectable. Conversely, a biopsy of the tumor is indicated under a questionable radiological diagnosis and/or in the case of locally advanced tumors requiring neoadjuvant therapy. 17
In localized disease, the technique of choice for histological diagnosis is through echoendoscopy. A CT-guided percutaneous biopsy can be considered only if the first option is not possible. In this case, special attention should be given to avoid tumor rupture or spillage. A biopsy is preferred over fine-needle aspiration to provide sufficient material to carry out a definitive histological and molecular diagnosis. 18 In patients with accessible metastatic disease, a CT- or ultrasound-guided biopsy can be preferred to approach the primary tumor.
Pathological features
Macroscopic characteristics
GISTs are variable in size, from centimetric lesions to big tumor masses greater than 20 cm. Typically, GISTs arise from the wall of the digestive tract and extend inward toward the mucosa, outward to the serosa, or in both directions. 19 Infrequently, they can ulcerate the overlying mucosa. Macroscopic areas of necrosis, hemorrhage, and cystic degeneration may be seen. 19 While sporadic GISTs grow as solitary masses, different patterns may raise the suspicion of particular entities, such as SDH-deficient GIST if multinodular, 11 and NF1 or familial GIST if multiple primary tumors are present. 20 Tumor rupture is an adverse prognostic factor irrespective of whether it takes place before or during surgery. 21
Histology
Three histological subtypes can be distinguished: spindle cell (77%), epithelioid (8%), and mixed (15%), with no prognostic significance.22,23 The epithelioid type is more frequent in the stomach and associated with PDGFRA mutations. 22 The mitotic count has prognostic value and should be expressed as the number of mitoses on a total area of 5 mm2. Strict criteria should be applied: pyknotic, dyskariotic, or apoptotic nuclei should not be regarded as mitosis. Due to intratumor heterogeneity, the assessment of risk stratification features such as mitotic count should be deferred to the surgical resection specimen, as a core biopsy can misguide its evaluation.
Immunohistochemistry
Over 95% of GISTs show expression of KIT (CD117) – cytoplasmatic, membranous, or perinuclear dot-like staining – regardless of the mutational status. Strong and diffuse staining is the most common pattern present in 75% of cases. In addition, GISTs can also express CD34 (70–90%), actin (20–30%), S-100 (8–10%), and desmin (2–4%).19,23 A small proportion of GIST (<5%) may show weak or no KIT expression, which is more commonly observed in PDGFRA-mutant GIST24,25 and dedifferentiated GIST. 26 ANO1/DOG1 immunostain is a sensitive and specific marker that is positive in approximately half of KIT-negative GISTs. 27 PDGFRA immunohistochemistry can also be useful in this population to predict the presence of PDGFRA mutations. 28
Differential diagnosis
The main differential diagnosis of spindle cell GIST comprises smooth-muscle tumors (leiomyoma and leiomyosarcoma), fibromatosis (desmoid), schwannoma, malignant peripheral nerve sheath tumor, inflammatory myofibroblastic tumor, solitary fibrous tumor, inflammatory fibroid polyp, and sarcomatoid carcinoma. The differential diagnosis of epithelioid GIST includes poorly differentiated carcinoma, neuroendocrine tumor, epithelioid leiomyosarcoma, PEComa, and malignant melanoma, among others. 29
Diagnostic evaluation: Final recommendations
An abdominal CT scan with contrast and image acquisitions of the arterial and portal phases is the first choice to study location and extension (IV,B).
Preoperative biopsy is necessary when there is a questionable radiological diagnosis and/or in the case of locally advanced or metastatic tumors (IV,C).
The pathology report must include information for risk assessment (anatomical location, tumor size, mitotic activity) and other important prognostic factors such as tumor rupture and margin status (Table 1) (III,A).
A basic immunohistochemistry (IHC) panel includes CD117, DOG1, actin, desmin, S-100, and CD34 (IV,B).
Required elements in the pathology report for resected GIST.

Tumor rupture is an additional adverse prognostic factor and should be recorded, regardless of whether it took place before or during surgery. These patients should be considered metastatic for treatment purposes.
The required total count of mitoses is per 5 mm2 on the glass slide section. In older microscopes, 50 HPF is equivalent to 5 mm2. Modern microscopes with wider 40× lenses/fields require approximately 20–25 HPF to encompass 5 mm2.
AJCC, American Joint Committe on Cancer; GIST, gastrointestinal stromal tumor; HPF, high-power fields; NGS, next-generation sequencing.
Molecular biology
The determination of GIST molecular profile is critical to tailor the use of the targeted agents approved against this disease. Approximately 85% of GISTs are driven by mutually exclusive activating mutations in KIT or PDGFRA. In addition, between 10% and 15% of GISTs are wild type (WT) for KIT and PDGFRA, and other events are involved in their pathogenesis 4 (Figure 1). Mutations in KIT and PDGFRA commonly cluster in well-known regions of these kinases and include a broad range of gain-of-function alterations such as deletions, missense mutations, duplications, insertions, and the combination of these. 30
KIT mutations
Between 75% and 80% of GISTs harbor primary clonal mutations in KIT, and the most commonly affected exons include 11, 9, 13, and 17. The majority of KIT mutations are found in exon 11 (~70%), which encodes the intracellular juxtamembrane domain. 4 Most of the alterations detected in exon 11 are deletions predominantly found between codons 550 and 579. Less frequently, missense mutations affect codons 557, 559, 560, and 576.31–33 KIT exon 9 codifies for the extracellular ligand-binding domain, is detected in 12–15% of cases, and is associated with small bowel location and greater malignant potential.30,33 These mutations commonly consist of duplications of codons 502 and 503. Finally, exons 13 and 17 codify the KIT ATP-binding site and the activation loop, respectively. They occur only as missense substitutions and are rather uncommon as primary mutations (<1%).4,30,33
Primary mutations in KIT are the initiating, clonal event present throughout the course of the disease. By contrast, secondary mutations in KIT emerge after the selective pressure exerted by TKIs and are associated with resistance in up to 90% of GIST patients. KIT secondary mutations cluster in the ATP-binding pocket (exons 13 and 14) and the activation loop (exons 17 and 18)34,35 (Figure 2).

KIT- and PDGFRA-mutant GIST.
PDGFRA mutations
The estimated frequency of PDGFRA mutations ranges between 5% and 10% and is associated with GIST of gastric location and epithelioid morphology. 36 Mutations are mainly found in the juxtamembrane (0.7%) and kinase domains (6%) encoded by exons 12 and 18, respectively. The D842V substitution in exon 18 is the most frequent PDGFRA alteration (65–75% of PDGFRA-mutated cases). Mutations in exon 14 are extremely infrequent (0.1%) 36 (Figure 2).
GIST KIT/PDGFRA wild type
KIT/PDGFRA WT GIST includes a hodgepodge of molecular alterations and resulting clinical phenotypes that need to be identified to predict the course of the disease, uncover inherited syndromes and, if indicated, tailor adequately specific treatments.
Succinate dehydrogenase-deficient GIST
The SDH protein complex is composed of four subunits: SDHA, SDHB, SDHC, and SDHD. The loss of any of these subunits through mutations or epigenetic silencing leads to the instability of the entire SDH complex, resulting in SDHB degradation and subsequent loss of its expression. For this reason, immunohistochemistry for SDHB is a diagnostic marker useful in the identification of this subgroup. 37 Overall, it is estimated that more than 80% of adult KIT/PDGFRA WT GIST are SDH deficient. 38
Clinically, these tumors tend to occur in the pediatric population (no sex predominance) and the young adulthood (with female predominance) and are restricted to the stomach. 11 SDH-deficient GISTs have characteristic morphologic features, including multinodular gastric wall involvement and occasional lymph node invasion. Microscopically, these GISTs feature epithelioid morphology, strong expression of KIT (CD117), and loss of SDHB expression in tumor cells. Although the clinical spectrum can be broad, progression is usually slow even after dissemination, and many patients can live years with metastases.37–39
Succinate dehydrogenase-competent GIST
Less than 20% of CD117-positive KIT/PDGFRA WT GIST are SDH competent, and therefore retain SDHB expression. This subgroup of tumors is characterized by other genetic abnormalities leading to mitogen-activated protein kinase pathway overactivation, such as NF1 loss-of-function mutations, BRAF V600E gain-of-function mutations, or chromosomal rearrangements involving the neurotrophic tyrosine kinase receptor (NTRK).12,38–40
NF1- and BRAF-mutant GISTs occur almost exclusively along the small bowel. However, while BRAF-mutant GISTs are isolated tumors, NF1-mutant GIST most commonly presents as a multifocal or multinodular disease. In this case, a previously unrecognized NF1-inherited syndrome should be excluded. 41 There are currently no specific morphological or IHC features, and the final diagnosis is only made through next-generation sequencing (NGS) panels.
Finally, comprehensive genomic profiling studies have identified a number of gene fusions in KIT/PDGFRA WT GISTs that involve chromosomal rearrangement involving the NTRK3, which represents an actionable alteration. 40 However, these cases seem to be exceptionally rare. 42 Given its rarity and the high false-positive rate for NTRK stain in non-NTRK-fused tumors, routine screening in GIST for NTRK abnormalities is not appropriate, and NGS panels can be used only if the conjoint assessment rises the diagnostic suspicion. 40
False-negative Sanger sequencing analysis
The sensitivity of routine Sanger sequencing is 20–25%. However, it is possible that the allele frequency of heterozygous KIT or PDGFRA mutations can be lower in tumor samples containing an abnormally elevated proportion of normal tissue or immune infiltrate. In our GEIS experience, 40–50% of KIT/PDGFRA WT GIST by Sanger sequencing turned out to be KIT- or PDGFRA-mutant when analyzed with NGS, which has a higher sensitivity owing to its ability to detect mutations at lower allele fractions of 1–5%. 43
Molecular biology: Final recommendations
Systematic molecular analysis during the diagnostic workup is strongly recommended for all GIST, given the relevant predictive and prognostic information provided. Genetic analysis also confirms the diagnosis of GIST in CD117/DOG-1-negative tumors (II,A).
It is advisable to centralize the mutational analysis in laboratories enrolled in an external quality assurance program and with expertise in GIST (IV,C).
The use of NGS panels is recommended in KIT/PDGFRA WT GIST without SDH deficiency (III,A).
The collection of fresh/frozen tissue is encouraged because new molecular pathology assessments can be made at later stages in the patients’ interest (V,C).
Risk stratification in localized GIST
Relapse risk assessment for primary GIST is paramount not only for providing prognostic information when trying to determine risk factors but also for estimating the potential benefit of adjuvant imatinib.
Risk stratification systems
The first proposed index (National Institute of Health – NIH Consensus or Fletcher et al. index) considered the size and mitotic count per 50 high-power fields (HPF) as the variables with independent prognostic value. 22 Miettinen and Lasota proposed a more complex index including primary tumor location after observing a lower recurrence rate in gastric GIST compared to other locations. Thus, the Armed Forces Institute of Pathology (AFIP)/Miettinen and Lasota risk criteria incorporated the anatomic site to tumor size and mitotic count. 44 Lately, the NIH consensus-modified criteria incorporated tumor rupture to these three prognostic factors, and it was the base for the risk estimation in the randomized trials studying adjuvant imatinib in localized GIST 45 (Table 2). Importantly, Miettinen considered a total area of 5 mm2 in 50 HPF characterized by the use of different optical components, while in practice 50 HPF typically corresponds to a total area of 10 mm2. Therefore, if Miettinen’s risk classification is used, the number of HPF equivalent to a surface area of 5 mm2 should be calculated based on specific microscope parameters.
Guidelines for risk assessment of primary GIST.

Source: Adapted with permission from Miettinen and Lasota. Copyright 2006 by Elsevier.
Defined as metastasis or tumor-related death.
Denotes small number of cases.
GIST, gastrointestinal stromal tumor.
The casuistry of the GEIS group showed that AFIP classification, unlike NIH risk criteria, exhibited statistical significance for distinguishing low-, intermediate-, and high-risk groups. 32 Other alternative risk classifications carry the limitations of categorized variables. To overcome this hindrance, Joensuu et al. proposed a risk classification based on the nonlinear modeling of mitotic count and size, analyzing them as continuous variables. The accuracy of this prediction was performed in a population-based registry, and generating heatmaps is superior to other risk classification models when the area under the curve was compared. The European Society for Medical Oncology guidelines are prone to use heatmaps as the best tool to offer risk estimation. Adjuvant imatinib is usually recommended for those cases with a probability of recurrence higher than 40% using the heatmaps model. 46
Molecular features predictive of risk prognosis
There is available evidence indicating that the type and location of the mutation influence the risk of recurrence. We found that deletions affecting codons 557 and/or 558 in KIT exon 11 (from now on, we will refer such deletions as ‘critical mutation’) have a higher recurrence risk within the first 3–4 years after surgery, 32 which was confirmed in larger series.47,48 A later retrospective study from the Conticanet network summarizing 1056 localized GIST cases found that intermediate-risk gastric cases harboring ‘critical mutations’ had a significant and independent worse prognosis. 49 Mutations within PDGFRA showed a trend toward a better prognosis.
Risk stratification: Final recommendations
We recommend the NIH consensus-modified risk criteria, as it has been widely used for the recommendation of adjuvant imatinib (III,A).
Deletion type of mutations affecting codons 557 and 558 confers a risk for recurrence regardless of its previous classification, according to different retrospective series (III,A).
Management of localized disease
Surgery
Complete surgical resection is the standard treatment for localized GIST. The goal is an R0 surgery with complete removal of the tumor, including an intact pseudocapsule. Recommended macroscopic resection margin is 1 cm. Tumor resection must be carefully performed to avoid tumor rupture.21,50 Peritoneal and hepatic surfaces should be carefully examined to exclude tumor spread. Lymphadenectomy is unnecessary except for SDH-deficient GIST, or if macroscopic lymph node involvement is detected. Segmental resection of the intestine and stomach (nonanatomical resection/‘wedge’ resection) is generally accepted, thus avoiding aggressive procedures with unnecessary removal of unaffected tissue. Radiological criteria for unresectability include infiltration of the celiac trunk, the superior mesenteric artery, or the mesenteric artery-to-portal vein. Occasionally, en bloc resection can be considered; however, multi-visceral resection should be avoided and multidisciplinary consultation with a GIST expert team is highly recommended.51,52
R1 resection of a GIST tumor neither involves higher risk of recurrence nor a worse survival, whereas a macroscopic incomplete resection (R2) is associated with worse outcome.53,54 Re-excision after R1 surgery is not well defined and might be offered if it does not imply major risks or functional consequences.
Laparoscopic resection should follow the same principles of open surgery, with the same goal of achieving an R0 resection.55,56 Overall, laparoscopic resection in GIST is not recommended in tumors >10 cm due to a high risk of tumor rupture. It can be considered in GIST <5 cm in favorable anatomical locations such as the greater curvature, the fundus, and the anterior gastric aspect. The extraction of the surgical specimen must be carried out in a bag to avoid locoregional dissemination. Nevertheless, the indication of laparoscopic resection should be agreed on a case-by-case basis following a multidisciplinary evaluation by teams with broad experience in laparoscopic surgery.
Small GIST < 2 cm
Esophagogastric or duodenal submucosal GISTs < 2 cm are primarily managed with endoscopic ultrasound. If a biopsy is feasible, the diagnosis of GIST should be made. If confirmed, endoscopic resection with complete excision and without tumor rupture can be an acceptable alternative to laparoscopic or open resections to minimize morbidity. Alternatively, patients can choose to undergo active surveillance. Likewise, if a biopsy is not feasible or the material is inadequate, the first option is normally active surveillance, although patients can opt for surgical or endoscopic resection. Initial follow-up with endoscopy can be performed every 3 months and can be prolonged in the absence of tumor growth. 17
Adjuvant treatment with imatinib
Although complete surgical resection is feasible in most localized GISTs, metastatic relapse occurs in approximately 40% of the patients. 46 Three phase III randomized clinical trials assessed the role of adjuvant imatinib to prevent disease recurrence and improve OS. ACOSOG Z9001 57 and SSGX-VIII/AIO 58 studies showed improved relapse-free survival (RFS) after 1 and 3 years, respectively, of adjuvant imatinib 400 mg daily. In addition, the SSGX-VIII/AIO trial demonstrated an increase in OS with 3 years of imatinib administration in comparison with 1 year in high-risk patients, according to the NIH-modified risk criteria. Lately, the EORTC 62024 phase III study randomized intermediate-risk and high-risk patients to 2 years of imatinib versus observation. 59 Despite the overt impact of imatinib on RFS, there were no differences in OS or in imatinib failure-free survival (IFFS) – an innovative end point capturing the time to the development of secondary resistance to imatinib in the metastatic setting – although there was a nonsignificant trend toward IFFS improvement in high-risk patients.
A more recent 10-year follow-up from the SSGX-VIII/AIO trial confirmed the long-term benefit of 3 years of adjuvant imatinib, showing 10-year OS rates of 79% compared with 65% in the 12-month group. 60 In view of these results, the scientific community agrees to recommend 3 years of adjuvant treatment with imatinib 400 mg daily in high-risk patients with imatinib-sensitive mutations in KIT or PDGFRA.
Two important questions remain open. First, the duration of adjuvant imatinib. Recently, the phase II, single-arm, PERSIST-5 clinical trial reported an estimated 5-year RFS of 90%, superior to historical data, although only 46 out of the 91 patients enrolled completed the 5 years of adjuvant imatinib. 61 The currently ongoing SSG XXII (NCT02413736) phase III trial randomizes surgically resected high-risk GIST patients to 3 versus 5 years of adjuvant imatinib. The second question relates to the potential benefit of adjuvant imatinib in particular situations, the most notable intermediate-risk patients with KIT primary mutations affecting 557 and/or 558 codons.32,48,49 Although molecular determinants of risk are not included in international consensus, based on the strong evidence reported by others and us, we recommend adjuvant imatinib in this subset of patients.
Adjuvant imatinib in special situations
- Tumor rupture, regardless of whether it occurs spontaneously or during the surgical procedure, is associated with a risk of relapse close to 100%. 21 Therefore, these cases should be considered metastatic and treated with imatinib accordingly. However, the total length of imatinib can be individually discussed in special situations such as tumor microperforation.
- The specific sensitivity of some KIT and PDGFRA primary genotypes should always be carefully considered for the indication of adjuvant imatinib. For instance, the benefit of the standard 400 mg dose has also been demonstrated retrospectively in KIT exon 9-mutant GIST patients, 62 a subset of GIST that benefits from a higher imatinib dose (see below). However, GIST with the primary PDGFRA D842V mutation and all GIST WT for KIT and PDGFRA are, by definition, insensitive to imatinib and thus adjuvant imatinib is not indicated.
- R1 surgery, by itself, does not constitute a criterion to weight in a potential use of adjuvant imatinib.
Neoadjuvant treatment with imatinib
A treatment strategy based on front-line imatinib followed by surgery can be occasionally indicated in GIST patients with locally advanced disease. Prior discussion in a multidisciplinary tumor board is mandatory. The final goal is to facilitate an R0 surgical procedure. Therefore, this approach can be attempted in those GISTs that are initially resectable, but at the expense of a mutilating surgery. 63 Typical examples of such scenarios are GIST masses in the gastroesophageal junction, duodenum, or rectum. Other locations can be considered as well, particularly in cases with a high risk of tumor rupture, that is, >10 cm necrotic masses. Molecular profiling before treatment initiation is critical since it guides dosing or discourages the use of imatinib.
Early response assessment, that is, after 4 weeks of treatment initiation is highly recommended since an unsuccessful imatinib treatment could impede a surgical procedure that otherwise could have been undertaken. Although a CT scan is sufficient for the evaluation of response, a PET/CT scan can be more advantageous to measure the efficacy of imatinib within a short period of time. 64 The recommended duration of neoadjuvant imatinib cannot be based on objective criteria. However, it is estimated that surgery could be performed between 6 and 12 months after initiation of imatinib, as maximal response and minimal risk of secondary resistance are expected in this time interval. 65 Imatinib can be stopped 24 h before the surgery and reinitiated once the oral tolerance has been confirmed.
Adjuvant treatment with imatinib can be indicated once the surgery is completed. To do so, a mitotic count should be taken from the tumor biopsy obtained prior to the neoadjuvant treatment. Likewise, tumor size and location will be taken from the baseline CT scan. If indicated, the total duration of preoperative and postoperative imatinib treatment should sum up the total of 3-year duration of a conventional adjuvant treatment.
Management of localized disease: Final recommendations
The standard treatment of localized GISTs is a complete surgical resection (III,A).
Adjuvant treatment with imatinib 400 mg daily is indicated in GIST patients at high risk of relapse-bearing imatinib-sensitive mutations in KIT or PDGFRA (I,A). Imatinib 400 mg daily is an option for patients with KIT exon 9 mutations (IV,C).
Molecular profile of KIT and PDGFRA is mandatory before initiation of adjuvant imatinib (II,A).
Treatment with neoadjuvant imatinib can be considered in a multidisciplinary tumor board in GIST patients with locally advanced disease in which tumor shrinkage can facilitate the surgical procedure (III,A).
Management of metastatic disease
First-line treatment with imatinib
Dose and efficacy
The standard dose of imatinib 400 mg per day 66 was established from two randomized phase III trials in metastatic GIST with positive immunostaining for CD117 (EORTC 62005 and NASG S0033).67,68 In both trials, daily doses of 400 versus 800 mg were comparable, obtaining a clinical benefit rate (complete response, partial response, and stable disease) of ~90%. The progression-free survival (PFS) in the EORTC trial favored the 800 mg dose, with a 2-year PFS of 52% versus 44% (HR 0.78), which was confirmed in a later meta-analysis. 69 Nevertheless, since this did not translate into survival advantage and lower doses were better tolerated, 400 mg daily is the standard recommended dose.
Predictive value of KIT and PDGFRA genotyping
The objective response rates for KIT exon 11, exon 9 mutants, and GISTs WT were 72%, 38%, and 28%, respectively, with a median time to progression (mTTP) of 25, 17, and 12,8 months, respectively.70–73 A later meta-analysis confirmed that only KIT exon 9-mutant GISTs benefit from imatinib 400 mg twice a day, thereby reducing by 42% the risk of progression and by 31% the risk of death. 69 Current data are insufficient to provide clear numbers for KIT exons 13 or 17, and therefore imatinib 400 mg is initially recommended.
Based on laboratory and scattered clinical data, the most common PDGFRA mutation in GIST, D842V, is deemed completely resistant to imatinib.36,74 By contrast, other mutations in PDGFRA not involving the D842V substitution appear to be sensitive.
Practical issues on first-line imatinib in metastatic patients
- How long should the therapy last? The BFR14 trial randomized patients with nonprogressive GIST to imatinib continuation versus interruption after 1, 3, or 5 years of treatment. Treatment interruption was consistently associated with disease progression even in patients with complete response. 75 Consequently, imatinib must be continued until disease progression or until unacceptable toxicity (I,A).
- Although seldom, some patients may experience imatinib intolerance. In this setting, treatment with second-line sunitinib should be discussed. Alternatively, nilotinib could also be contemplated (II,B). 76
- Plasma levels can potentially guide treatment decisions in patients with unexpected poor efficacy with imatinib, based on retrospective data correlating low plasma levels (<1110 ng/mL) with shortened mTTP (IV,B). 77
- Metastatic relapse after finalization of adjuvant imatinib does not usually harbor acquired resistance. Therefore, the recommendation is to reintroduce imatinib using first-line doses (IV,A).
Treatment for patients with disease progression following imatinib failure
Imatinib dose escalation
If compliance is correct, systemic therapy should be changed. One option to consider is to increase the dose to 400 mg twice daily. A total of 30% of the patients that crossed-over from 400 to 800 mg in the EORTC 62005 and the NASG S0033 trials67,78 achieved disease control. Although the mTTP was modest, 81 days, 18% of the patients remained free of progression 1 year after cross-over. Patients with KIT exon 9 mutation appear to benefit particularly from this approach, whereas it is more limited in KIT exon 11 mutants.79–81 The incidence of anemia and asthenia increases significantly with this dosage; therefore, a strict follow-up is required.67,68
Second line: Sunitinib
The multikinase inhibitor sunitinib is an equally valid alternative to high-dose imatinib after progression to imatinib 400 mg. Sunitinib showed improvement in the mTTP from 1.5 to 6.3 months in comparison to placebo. 82 The recommended dose is 50 mg orally once daily over 4 weeks followed by a 2-week rest period. A later single-arm phase II trial with a continuous daily dose of 37.5 mg showed comparable activity and better tolerability, thus constituting a valid alternative. 83 The most common side effects were asthenia, skin toxicity, diarrhea, hypertension, and hypothyroidism. Specific molecular backgrounds benefit particularly from sunitinib treatment: KIT exon 9 primary mutation, SDH-deficient GIST, and secondary mutations in KIT exons 13 and 14.11,84,85
Sunitinib remains the standard second-line treatment after the results of the INTRIGUE trial, in which sunitinib and ripretinib showed comparable outcomes, with a PFS of 8.3 and 8.0 months, respectively. 86
Third line: Regorafenib
The multikinase inhibitor regorafenib is approved at the doses of 160 mg daily, 3 weeks on, 1 week off, as the third line of treatment based on the results of the phase III GRID trial, 87 exhibiting a PFS of 4.8 months compared with 0.9 in the placebo arm. The most common toxicities are hypertension, hand–foot skin reaction, and diarrhea. In contrast to sunitinib, regorafenib appears to be more active against secondary KIT mutations located in the activation loop (exons 17 and 18). 35
Regorafenib remains the standard third-line treatment after the results of the VOYAGER trial, in which regorafenib and avapritinib showed comparable outcomes, with a PFS of 5.6 and 4.2 months, respectively. 88
Fourth line and beyond
Ripretinib is a type II TKI that antagonizes both the juxtamembrane domain and the activation loop of KIT and PDGFRA. The phase III INVICTUS trial proved that ripretinib 150 mg daily was superior to placebo after progression to all standard treatments, showing a PFS of 6.3 months compared to 1.0 month. 89 The toxicity profile of ripretinib is comparable with imatinib, albeit with a higher incidence of alopecia and mild hand–foot syndrome.
Pazopanib 90 and cabozantinib, 91 among other multi-targeted TKI, 39 have shown some interesting activity in imatinib-resistant patients in phase II clinical trials, although large randomized trials are lacking and they are not approved for the treatment of GIST. However, they might be used as compassionate use in selected patients after progression to all standard treatments. Finally, continuous TKI suppression after progression to a given agent appears to improve the outcomes of metastatic GIST patients, as it has been demonstrated with imatinib rechallenge 92 and the maintenance of sunitinib or avapritinib beyond progression.93,94 Therefore, given the rapid progression of TKI-refractory GIST patients, 89 we recommend maintaining or re-challenging a prior TKI while a therapeutic alternative is available.
PDGFRA D842V-mutant GIST patients
The substitution of aspartic acid by valine at codon 842 in PDGFRA exon 18 (D842V) occurs in ~5% of all GIST and is known to be resistant to all therapeutic agents.36,74 The activity of avapritinib, a type I TKI, was studied in the phase I NAVIGATOR trial in 56 D842V-mutant GISTs, including 11 TKI naïve. The overall response rate was 91%, clinical benefit rate 98%, and median PFS 34 months, which constitutes an unprecedented activity in this molecular subset of GIST.95,96 Most common toxicities are nausea, fatigue, anemia, diarrhea, and edema, and also a characteristic increase in cognitive effects in 37% of the patients that require strict monitoring. 97
The activity of all approved agents for the treatment of GIST is summarized in Table 3.
Updated activity of approved tyrosine kinase inhibitors in advanced/metastatic GIST.

GIST, gastrointestinal stromal tumor; mo, months; mPFS, median progression-free survival; N.A., not available; ORR, overall response rate; SD, stable disease (at least 12 weeks).
Local treatment of metastatic GIST
Several retrospective series, including ours, have consistently demonstrated that cytoreductive surgery aiming at R0/R1 disease following initial response to imatinib is associated with improved long-term survival.98,99 However, incomplete resection, including debulking surgery, does not seem to prolong survival.98,100 The absence of data discourages the combined use of hyperthermic intraperitoneal chemotherapy (HIPEC). Together, and despite their retrospective and likely biased nature, these works support that selected patients may undergo cytoreductive surgery after initial response to imatinib and if the metastatic disease is deemed resectable. Given their metastatic nature, it is important to avoid mutilating procedures and maintain imatinib afterward.
While all these studies do not recommend surgery in patients with multifocal progression, they agree that the resection of unifocal/limited progression may improve PFS.98,100,101 This decision should be taken on an individual basis after discussion in a multidisciplinary tumor board. Seemingly, this approach benefits mainly patients on imatinib. However, it can be considered in later lines depending on the time of the drug. As in patients with cytoreductive surgery, the same TKI must be continued after resection. Finally, the evidence for other approaches such as embolization or radiofrequency is lacking, and therefore surgery should be prioritized.
Imatinib for metastatic disease: Final recommendations
Genotype is mandatory for treating advanced/metastatic GIST patients (II,A).
Imatinib 400 mg daily is the recommended dose in the first line (I,A).
Imatinib 400 mg every 12 h is the recommended dose for GIST with KIT exon 9 mutation (II,A).
It is unclear whether imatinib should be the first line in GIST KIT/PDGFRA WT (IV,C).
Debulking surgery aiming an R0/R1 surgery can be considered in selected patients after initial response to imatinib (IV,B).
Imatinib-resistant disease: Final recommendations
Confirm adherence to treatment and rule out drug interactions at the time of progression to any TKI (III,B).
After the failure of imatinib, the standard second-line treatment is sunitinib 50 mg daily 4/2 (I,A) or 37.5 mg continuously (III,C).
Before sunitinib, imatinib dose escalation to 400 mg twice daily can be considered, particularly in patients with KIT exon 9-mutant GIST (III,B).
Standard third- and fourth-line treatments are, respectively, regorafenib 160 mg daily 3/1 (I,A) and ripretinib 150 mg once daily (I,A).
Avapritinib 300 mg daily is the only effective treatment available for PDGFRA D842V-mutant GIST and it should be introduced, if possible, as the first line (III,A).
Maintenance of TKI pressure improves outcomes and it is advised while an alternative therapeutic option is unavailable (III,B).
Surgery of unifocal/limited progression can be considered after discussion in a multidisciplinary tumor board (IV,C).
Response evaluation and follow-up
Localized resectable disease
There are no studies analyzing the efficacy of different follow-up strategies. Most accepted recommendations advocate adjustment to the risk of recurrence with time based on known risk factors. 17 We suggest the following schedules according to each risk group for 10 years: 46
- Very low risk. If surgically removed, no follow-up.
- Low risk. Annual CT scan.
- Intermediate risk and high risk. 1–2 years CT scan every 4 months; 3–5 years every 6 months; annually thereafter. Note that once imatinib is withdrawn, relapses occur most frequently within the following 2 years.
Unresectable or metastatic disease
Follow-up should be conducted every 3 months from the beginning and can be prolonged up to every 6 months if the response is obtained, especially if response remains beyond a 5 years. Modified RECIST 1.1 and Choi criteria must be considered to avoid confusion with pseudoprogression.
Conclusions and future perspectives
Since the last update of the GIST GEIS guidelines in 2017, two new TKIs, ripretinib and avapritinib, have been approved by the regulatory authorities for the treatment of metastatic GIST. Two later phase III randomized trials have helped in consolidating sunitinib and regorafenib as the standard-of-care second and third lines, respectively. Given the continuous understanding of GIST biology and the high potential to develop international randomized trials, it is likely that the therapeutic field will be shaken in the years to come. In this sense, we encourage to treat GIST patients at any stage in clinical trials to boost drug development.
Two more aspects of GIST management will be clarified over the next years: First, whether 5 years of adjuvant imatinib is superior to 3 years is the current standard second, the role of circulating tumor DNA in taking treatment decisions, particularly in metastatic GIST. Finally, we want to emphasize that, despite all this therapeutic success, GIST is a rare tumor. As such, the collective evidence has consistently demonstrated that the treatment of these patients in sarcoma referral centers improved their outcomes. Therefore, we favor a continuous dialogue between patients’ physicians from home and sarcoma expert institutions to have GIST patients referred in their best interest.
Footnotes
Acknowledgements
We appreciate the Spanish Sarcoma Research Group (GEIS) for its administrative support.