
Abstract
Since its approval by the US Food and Drug Administration in February 2002, the tyrosine kinase inhibitor, imatinib, has become the standard of care for patients with metastatic or unresectable KIT-positive gastrointestinal stromal tumors (GISTs). Imatinib functions by blocking the adenosine triphosphate binding site of the constitutively activated mutant KIT or platelet-derived growth factor receptor α, effectively shutting down the oncogenic signal that drives up to 90% of these tumors. In doing so, it has transformed the management of a condition previously refractory to systemic treatments and established GIST as a model for the use of targeted therapies and oncogene addiction in solid tumors. However, while more than 80% of patients will receive clinical benefit from imatinib monotherapy, more than half will develop progressive disease by 2 years. In this article we review the mechanism and patterns of imatinib resistance in GIST; attempt to offer a practical schema for managing imatinib-refractory patients; and lastly, offer some insight as to future directions and emerging therapeutics for the management of this highly interesting and challenging disease.
Keywords
Introduction
Gastrointestinal stromal tumors (GISTs) are rare mesenchymal tumors of the digestive tract that most commonly arise in the stomach (60–65%) or small intestine (25–30%) [Miettinen and Lasota, 2006a]. They are believed to derive from the interstitial cells of Cajal, with both sharing almost universal expression of the receptor tyrosine kinase, KIT. Activation of the mitogen-activated protein kinase (MAPK) and phosphatidiyl-3-kinase (PI3K) pathways appears central to GIST pathophysiology. Mutually exclusive activating mutations in the upstream tyrosine kinase receptors KIT and platelet-derived growth factor receptor α (PDGFRA) are identifiable in 80–85% and 5–10% of cases respectively. Over- expression of insulin-like growth factor receptor (IGFR) or direct mutation of BRAF are amongst an ever-increasing range of genotypic changes identified that have been implicated in subsets of KIT/PDGFRA wild-type tumors [Tarn et al. 2008; Agaimy et al. 2009].
Whilst small GISTs are often asymptomatic and detected during investigations or surgical procedures for unrelated causes, larger tumors present with nonspecific symptoms such as gastrointestinal (GI) blood loss, abdominal pain, bloating or early satiety. Localized GISTs are best managed with surgical resection. However, up to 50% will recur, with the risk being most strongly related to tumor size, mitotic activity and site along the GI tract [Fletcher et al. 2002; Miettinen and Lasota, 2006b]. Unresectable or metastatic GIST is considered to be chemotherapy resistant. Response rates to conventional chemotherapy agents either individually or in combination are generally less than 10%[Dematteo et al. 2002] and the median survival for patients with metastatic GIST in the chemotherapy era has been estimated at between 15 and 19 months [Mudan et al. 2000; Gold et al. 2007; Agaram et al. 2008].
The identification of KIT and subsequently PDGFRA-activating mutations in the majority of GISTs has transformed the systemic management of this disease [Hirota et al. 1998; Heinrich et al. 2003b]. Imatinib mesylate (Gleevec; Novartis, Basel, Switzerland) is an orally active small molecule inhibitor of type III receptor tyrosine kinases, including KIT and PDGFRA. It binds competitively to the adenosine triphosphate (ATP) binding site of these kinases, thereby preventing their activation and the subsequent phosphorylation of downstream activating partners. Since its approval by the US Food and Drug Administration (FDA) in 2002, it has become the first-line standard of care for patients with unresectable or metastatic GIST. Most patients can expect to benefit. Response rates are between 40% and 50% and a further 20–30% of patients will have prolonged disease stabilization with a median progression-free survival (PFS) of just under 2 years (Table 1).
Key trials of imatinib in advanced c-KIT-positive gastrointestinal stromal tumors.
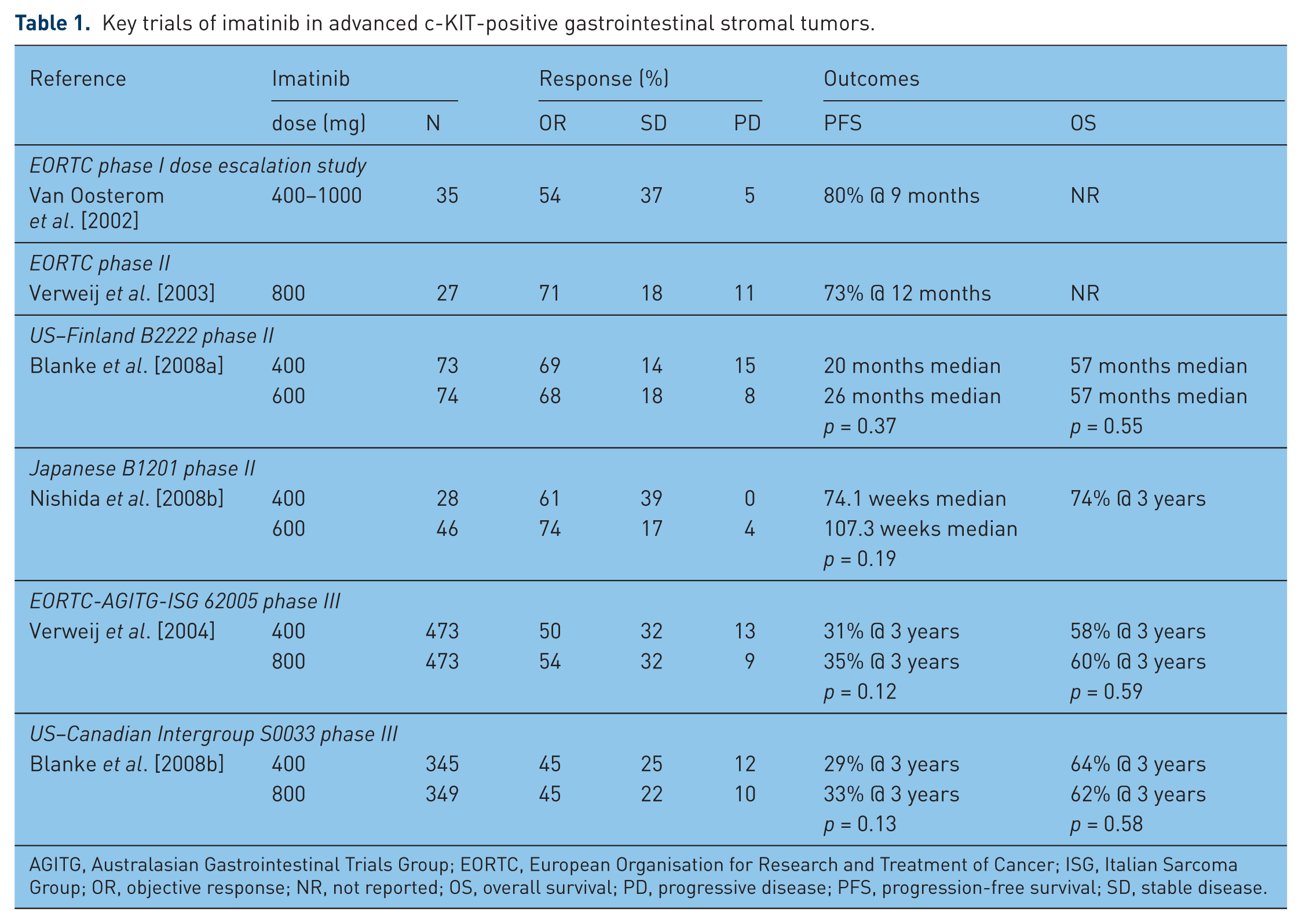
AGITG, Australasian Gastrointestinal Trials Group; EORTC, European Organisation for Research and Treatment of Cancer; ISG, Italian Sarcoma Group; OR, objective response; NR, not reported; OS, overall survival; PD, progressive disease; PFS, progression-free survival; SD, stable disease.
Molecular predictors of response to imatinib
Unfortunately, the majority of patients will eventually develop disease resistant to imatinib therapy. The underlying KIT or PDGFRA mutation has been identified as the strongest predictor of imatinib sensitivity [Heinrich et al. 2003a; Debiec-Rychter et al. 2004]. Mutations are known to cluster in certain genetic hotspots that result in functional activation of the resultant protein receptor [Corless et al. 2004]. Primary oncogenic KIT mutations occur most commonly in exon 11 (57–80%) which encodes the intracellular juxtamembrane domain and are followed by mutations of the extracellular domain located within exon 9 (5–18%). Rare primary mutations have been identified within the split kinase domain, represented by exons 13 (encoding the ATP-binding pocket) and exons 17 (which encodes the activation loop). Structurally analogous mutations occur in the PDGFRA gene exons 12, 14 and 18 (corresponding to KIT exons 11, 13 and 17) although the distribution of mutations is different, with PDGFRA exon 18 activation loop mutations most common (>90%) (Figure 1).

Frequency of primary KIT and platelet-derived growth factor receptor α (PDGFRA) mutations in gastrointestinal stromal tumors.
In the phase II and III imatinib clinical trials, patients with KIT exon 11 mutations demonstrated a higher response rate and longer survival outcomes than those with exon 9 or no detectable mutation [Heinrich et al. 2003a; Debiec-Rychter et al. 2006; Heinrich et al. 2008b]. GISTs with KIT exon 9 mutations appear to be more dose dependent. A meta-analysis of the Euro-Australian 62005 and North American S0033 phase III trials, which randomized patients with advanced GIST between initial standard dose (400 mg daily) imatinib or high-dose (800 mg daily) imatinib, demonstrated a higher response rate (47% versus 21%, p = 0.0037) and longer PFS (p = 0.017) for patients with KIT exon 9 mutations when treatment commenced at the higher dose. Although there was a trend to overall survival benefit, this was not statistically significant (p = 0.15), presumably due to allowed crossover. These findings were not replicated with any other GIST genotype [Meta-GIST, 2010]. Furthermore, imatinib appears to be only occasionally active against KIT and PDGFRA ATP-binding pocket mutations (e.g. KIT exon 13 K642E mutations are associated with response, whereas exon 13 V654A mutations are resistant to imatinib), but is generally considered inactive against the most common PDGFRA exon 18 D842 activation loop mutation [Frost et al. 2002; Corless et al. 2005; Heinrich et al. 2008b; Mcauliffe et al. 2008]. These findings are best explained by considering the mechanism of action of imatinib, which binds most avidly to the inactivated conformation of the ATP-binding pocket. Direct mutation of this site, or secondary structural changes to the active form caused by activation loop mutations, result in a decreased binding affinity for imatinib.
Imatinib resistance mechanisms
Intrinsic (or primary) imatinib resistance is therefore most commonly related to the primary GIST genotype. Clinically, 10–15% of patients will have primary resistant GIST, defined as an absence of objective response or disease stabilization lasting less than 3–6 months. Most of these patients will have imatinib-resistant KIT exon 9 or PDGFRA exon 18 D842V mutations, or no detectable mutation at all [Heinrich et al. 2003a, 2008; Debiec-Rychter et al. 2004].
Acquired (or secondary) resistance is seen in initially responding or stable GIST and develops at a median time of 18–24 months. The most commonly identified mechanism is the emergence or acquisition of secondary KIT mutations in exons 13, 14 or 17. Secondary mutations have been identified in 40–80% of tumor biopsy samples obtained from patients progressing on imatinib and are more common when the patient has a primary KIT exon 11 mutation [Antonescu et al. 2005; Heinrich et al. 2006; Wardelmann et al. 2006; Desai et al. 2007; Nishida et al. 2008a]. Furthermore, when multiple tumor samples have been available from a single patient, polyclonal resistance mechanisms are commonly identified. Coexisting distinct resistance mutations at an inter-lesional and intra-lesional level have been demonstrated to occur in as many as two-thirds of tested patients [Liegl et al. 2008]. This high degree of genetic heterogeneity is likely to be a significant limiting factor in attempts to develop effective single agent therapies for imatinib-resistant GIST. Other identified mechanisms of acquired resistance have included amplification of KIT [Debiec-Rychter et al. 2005; Miselli et al. 2007] and pharmacokinetic resistance that may involve altered activity of drug transporters, induction of the cytochrome P450 (CYP)3A4 isoenzyme, and poor patient compliance [Eechoute et al. 2011].
Management of imatinib-resistant or intolerant GIST
Confirm radiological progression
GISTs treated with imatinib do not always follow conventional patterns of response or progression. It is therefore important that patients are continued on imatinib until true progressive disease is confirmed. While a decrease in tumor volume on computed tomography (CT) imaging is the most common response to imatinib treatment, responding tumors may occasionally increase in size, a result of intratumoral hemorrhage or myxoid degeneration; or apparently new hypodense liver lesions may manifest, as treated tumors become less vascular and better delineated against normal background structures [Choi et al. 2004; Linton et al. 2006]. These radiographic features are most common in the first few months of imatinib therapy and are often observed in the presence of other responding lesions or an improvement in patient symptoms supporting a beneficial treatment response. True early progression is most commonly homogenous – consistent with known mechanisms of primary resistance.
In contrast, the most common pattern of delayed progression is the development of a new nodule within a pre-existing and responding tumor – commonly without any significant increase in total tumor volume [Shankar et al. 2005]. Such a pattern of progression is consistent with the outgrowth of a resistant clone following the acquisition of secondary mutations [Desai et al. 2007]. Other more conventional patterns of progression are also seen, and progression can be focal, multifocal or global. Fluorodeoxyglucose positron emission tomography (FDG-PET) imaging to assess the metabolic response of GISTs to therapy and detect early signs of reactivation is valuable when doubt exists based on conventional imaging. It may also be useful in identifying patients with isolated progression who may benefit from local therapies [Goerres et al. 2005].
Surgical resection or ablation of focally progressive GIST
Patients who are medically fit with surgically accessible focally progressive disease should be considered for resection. The rationale behind this approach is the elimination of drug-resistant clones that will allow ongoing therapy with imatinib, and the preservation of future systemic strategies for later in the disease course.
In a number of case series from different institutions, carefully selected patients with focally progressing lesions have achieved durable median PFS intervals of between 8 and 12 months following limited resection [Raut et al. 2006; Al-Batran et al. 2007; Dematteo et al. 2007; Mussi et al. 2010]. When surgery may not be possible, limited evidence exists that similar benefits could be obtained with nonsurgical ablative techniques such as radiofrequency ablation or embolization [Dileo et al. 2004; Kobayashi et al. 2009]. In contrast, for patients with generalized progression, surgery is unlikely to offer any significant benefit as most will progress again within 3 months and overall survival is usually short [Raut et al. 2006; Dematteo et al. 2007].
Drug exposure and imatinib dose escalation
Inadequate drug exposure is likely to play a role in progressive disease despite standard dose imatinib in up to 30% of patients. A correlation between low trough plasma levels of imatinib and poorer GIST outcomes was demonstrated in the pharmacokinetic substudy of the phase II B2222 trial [Demetri et al. 2009b]. In this study, steady state imatinib concentrations in the lowest population quartile were associated with a shorter time to progression (11.3 months versus >30 months, p = 0.0029) and lower overall response rate (44.4% versus 69.1%, p = 0.0601). A similar relationship between plasma drug levels and response was reported in a smaller population-based study that also identified a link between GIST genotype and drug level sensitivity [Widmer et al. 2010]. In this study the response rates of GISTs with KIT exon 9 mutations were shown to be more dependent on achieving adequate drug exposure than those with KIT exon 11 mutations – consistent with the preclinical data demonstrating a 5–10-fold higher inhibitory concentration for this mutation in cell line assays [Heinrich et al. 2008b].
In these and other studies significant interpatient variability in imatinib steady state drug levels has been demonstrated. Independent of drug dose, variations in imatinib plasma concentrations have been associated with various host factors, such as patient weight, protein binding (albumin, α1 acid glycoprotein), white cell count, prior gastrectomy and CYP activity [Gambacorti-Passerini et al. 2003; Larghero et al. 2003; Yoo et al. 2010]. Certain patient controllable factors can also significantly reduce circulating imatinib plasma levels and should be excluded in any patient with progressive disease. Drug interactions with potent CYP3A inducers, such as enzyme-inducing antiepileptic drugs or rifampicin, can reduce plasma imatinib concentrations by up to 74% [Bolton et al. 2004; Wen et al. 2006] and potentially impact on treatment efficacy. Poor treatment compliance is also recognized as a significant issue for patients on long-term imatinib therapy. For instance, a prescription-filling audit of over 4000 patients on imatinib for chronic myeloid leukemia or GIST identified a compliance rate of only 75%, with total compliance being achieved by less than half of patients [Tsang et al. 2006].
In patients on the standard 400 mg daily dose of imatinib in whom noncompliance and drug interactions have been ruled out, dose escalation to 800 mg per day may achieve sufficient plasma drug levels to re-establish disease control. Patients who crossed over to the higher 800 mg daily dose on progression in the Eastern Cooperative Oncology Group (ECOG) 62005 trial achieved clinical benefit in 29% of cases. Although the median PFS was short lived at 81 days, 18.1% of patients maintained disease control beyond 12 months [Zalcberg et al. 2005]. Similar outcomes were reported from the S0033 study – with 36% of patients benefiting for a median duration of 4 months [Rankin et al. 2004]. Dose escalation in selected patients already receiving imatinib at 400 mg daily is generally well tolerated, although an increased incidence in anemia and fatigue has been reported.
Second-line sunitinib for imatinib-resistant or -intolerant gastrointestinal stromal tumor
Sunitinib malate (Sutent, SU11248; Pfizer, New York, USA) is an orally active multitargeted tyrosine kinase inhibitor with activity against the KIT, PDGFR, vascular endothelial growth factor receptor (VEGFR), RET and FMS-like tyrosine kinase receptor 3 (FLT-3). Similar to imatinib, sunitinib binds to the inactive conformation of the ATP-binding pocket and blocks receptor tyrosine kinase auto activation [Gajiwala et al. 2009]. However, in comparison to imatinib, its structure and smaller molecular size affects its selectivity to this site, contributing to its broader spectrum of activity and toxicity profile. Sunitinib has in vitro and clinical activity against imatinib-resistant KIT exon 13 and 14 mutations affecting the ATP-binding pocket, as well as greater activity against wild-type KIT and exon 9 mutations [Prenen et al. 2006; Heinrich et al. 2008a; Gajiwala et al. 2009]. Secondary KIT exon 17, and the analogous PDGFRA exon 18 mutations, affect the activation loop and cause a change in structure of the ATP-binding pocket to the active conformation – they are resistant to imatinib and sunitinib.
The benefit of sunitinib in patients who are intolerant to imatinib or who have imatinib-refractory GIST has been established through a series of clinical trials. The phase I/II trial established an intermittent dosing schedule of 50 mg daily for 4 weeks on with 2 weeks off as an appropriate maximum tolerated dose [Maki et al. 2005]. The follow-up phase III trial randomized patients (2:1) between this dose and placebo, with the option to crossover on progression [Demetri et al. 2006]. Patients who received sunitinib upfront had a significant fourfold increase in time to progression (27.3 versus 6.4 weeks, p < 0.0001), which translated into an improvement in overall survival [hazard ratio (HR) 0.49, p = 0.007] despite the study’s crossover design. Based on these results, sunitinib was approved by the FDA in 2006 for patients with advanced GIST who are intolerant or resistant to imatinib.
Translational studies have helped to identify patients most likely to benefit from sunitinib (Table 2). Pre- and post-imatinib biopsies were available for the majority of patients on the phase I/II trial and have been correlated against outcomes [Heinrich et al. 2008a]. In keeping with its known preclinical activity, benefit (defined as a response or stable disease lasting longer than 6 months) was observed in all major primary KIT genotypes: KIT exon 9 (58%), KIT exon 11 (34%), and wild-type KIT/PDGFRA (56%). Secondary mutations were identified in 33% of patient samples and were significantly more common in patients with primary KIT exon 11 mutations than those with exon 9 mutations (73% versus 19%, p = 0.0003). Consistent with prior imatinib studies, these hotspots clustered around the ATP-binding pocket (exons 13 and 14) and the activation loop (exon 17). When present these mutations heavily influenced the likelihood of benefit from sunitinib with patients with sensitive KIT exon 13/14 mutations having a significantly longer PFS than those with sunitinib-resistant KIT exon 17/18 mutations (7.8 versus 2.3 months, p = 0.0157). Molecular analysis from the phase III study has not yet been presented. However, in this study it was noted that patients enrolled because of intolerance to imatinib appeared more likely to achieve a partial response than those with imatinib resistance [Demetri et al. 2006]. Associated substudies also identified two novel methods of predicting benefit for patients already on treatment. Both a decrease in circulating levels of soluble fragment of KIT (sKIT) at 12 weeks, and a metabolic response on FDG-PET at 4 weeks correlated strongly with a longer median time to progression [Deprimo et al. 2009; Prior et al. 2009].
Clinical benefit of sunitinib in the phase I/II trial in imatinib refractory gastrointestinal stromal tumor by primary and secondary tumor genotype.

Clinical benefit, response or stable disease for ≥ 6 months.
PDGFRA, platelet-derived growth factor receptor α; WT, wild type.
In the phase III study, sunitinib was associated with serious adverse events in roughly 20% of patients. Most commonly reported were fatigue, hand–foot syndrome, asthenia and hypertension. More recently, it has been associated with thyroid dysfunction, and clinical hypothyroidism may account for many of the nonspecific symptoms associated with its use [Desai et al. 2006; Mannavola et al. 2007]. Monitoring of thyroid function, and hormone replacement therapy when appropriate, may reverse many of these symptoms. Patients will occasionally experience tumor flare symptoms that relate to sunitinib’s intermittent dose scheduling. In part to address this issue, a phase II trial of sunitinib 37.5 mg with continuous daily dosing was performed and demonstrated retained activity and tolerability [George et al. 2009]. The published median PFS of 34 weeks compared favorably with that achieved in the phase III trial of sunitinib dosed with the intermittent dose schedule (27 weeks). Based on these results, and in the absence of any direct head-to-head comparisons, many GIST experts have now adopted this simpler, continuous dosing schedule.
Emerging strategies for managing imatinib and sunitinib-resistant gastrointestinal stromal tumor
Currently sunitinib remains the only approved agent for the treatment of imatinib-resistant or -intolerant GIST. However, with only half of patients likely to benefit and a median PFS of just over 6 months it is clear that alternative second-line and active third-line therapies are urgently required. The problem of treating advanced GIST post imatinib therapy is substantially more complex than that in the first-line setting. The diverse heterogeneity of resistance mechanisms, already described, which includes primary resistant genotypes, multiple and coexisting secondarily acquired resistance mechanisms, as well as any pharmacokinetic alterations, is a challenge for drug development and clinical trial design.
An unselected compound needs to have activity against all of the more commonly encountered resistance genotypes to consistently achieve durable responses. Therefore, it is not surprising that sunitinib, which lacks significant activity against KIT and PDGFRA activation loop mutations, has relatively limited efficacy. Potential strategies to overcome this problem include next generation broad activity tyrosine kinase inhibitors; targeting downstream signaling pathways (MAPK, PI3K); or novel compounds that can inhibit TKR activity independent of specific mutations. Personalizing drug selection based on secondary mutation analysis is likely to be confounded by the high frequency of demonstrated coexisting acquired mutations following imatinib therapy. However, it may be beneficial to stratify or select patients based on primary mutations – particularly PDGFRA mutant and KIT/PDGFRA wild-type tumors – and the molecular analysis of tumors from patients treated on the phase I/II sunitinib trials suggests predicting outcome based on known secondary GIST mutations is of at least some merit [Heinrich et al. 2008a]. Drug combinations may be necessary to most effectively suppress coexisting resistant clones, with the transition of well tolerated effective regimens into the first-line setting to delay their initial emergence. It is also likely to be important to maintain effective direct inhibition of KIT, both in combination strategies and in the comparator arms of phase III trials, due to likely persistence of cell populations that remain responsive to KIT inhibition in progressing disease, and the possibility of a tumor flare phenomenon on KIT inhibitor withdrawal [Bono et al. 2004].
ATP competitive tyrosine kinase inhibitors
Until recently, nilotinib represented the lead compound amongst a vast array of orally active multikinase inhibitors undergoing clinical development for advanced GIST (selected compounds summarized in Table 3). In preclinical studies nilotinib demonstrated activity against imatinib-resistant KIT exon 11 and 13 (V654A) or 17 (D820G, N882K) double mutants [Roberts et al. 2007; Guo et al. 2009; Cullinane et al. 2010] and favorable pharmacokinetics, achieving 7- to 10-fold higher intracellular drug concentrations than imatinib [Prenen et al. 2006]. However, despite promising results in initial patient trials, it has failed to demonstrate significant clinical benefit in phase III trials – either as a third-line agent, or in first-line therapy – where after the initial interim analysis it was felt to be unlikely to demonstrate superiority to imatinib leading to the trial’s termination and the decision to discontinue further development of nilotinib in GIST [Reichardt et al. 2010]. Sorafenib, and the structurally related regorafenib, are promising compounds that have superior in vitro activity to imatinib against KIT exon 9 mutants; and common secondarily acquired KIT mutations, including exon 14 gate keeper mutations (T670I) and activation loop mutations [Wilhelm and Chien, 2002; Guo et al. 2007; Wilhelm et al. 2011]. Both compounds are also potent inhibitors of angiogenesis and MAPK signaling through inhibition of VEGFR and RAF kinases respectively. Regorafenib in particular, has been demonstrated to have a potential role in GIST patients refractory to standard therapies. The GRID (GIST – Regorafenib In Progressive Disease) trial, a phase III study of third- or forth-line regorafenib versus placebo, was recently reported. Patients on regorafenib achieved a superior PFS of 4.8 months versus just 0.9 months for those receiving best supportive care and placebo (HR 0.27, p < 0.0001). An overall survival benefit was not demonstrated, although 85% of patients randomised to placebo had crossed over to regorafenib following progression [Demetri et al. 2012].
Selected agents under investigation for the management of advanced gastrointestinal stromal tumors.

BSC, best supportive care; DCR, disease control rate; HSP-90, heat shock protein-90; IM, imatinib; IM-INT, imatinib intolerant; IM-RES, imatinib resistant; ITT, intention to treat; mTOR, mammalian target of rapamycin; NCT, national clinical trial number; NE, not evaluable; NI, nilotinib; OS, overall survival; PD, progressive disease; PDGFR(A), platelet-derived growth factor receptor (α); PFS, progression free survival; PR, partial response; SD, stable disease; SU, sunitinib; TTP, time to progression; VEGFR, vascular endothelial growth factor receptor.
Source: www.clinicaltrials.gov (accessed 25 January 2012).
Non-ATP competitive targeting of TKR function
Heat-shock protein 90 (HSP90) is a chaperone protein that appears to be particularly important for the stability of mutant oncoproteins [Trepel et al. 2010]. Inhibitors of HSP90 may therefore provide a means of targeting oncogenic KIT or PDGFRA in a way that is independent of the underlying primary or secondary mutations [Bauer et al. 2005]. Unfortunately IPI-504, the first orally active HSP90 inhibitor, failed to demonstrate significant clinical activity in a phase III trial before it was ultimately discontinued due to an excess of unexpected deaths in the study treatment arm [Demetri et al. 2010]. Nevertheless other drugs in this class, such as STA-9090, remain in clinical development and promise less toxicity and increased potency from GIST preclinical models [Wang et al. 2010]. A novel class of direct tyrosine kinase inhibitors, the switch pocket kinase inhibitor (SPKI), offers an alternative means of overcoming multiple resistant genotypes. The switch pocket is a region adjacent to the ATP-binding site on KIT and other kinases that binds the activation loop (the so-called switch) when the kinase is active. By competing for this region, SPKIs prevent the kinase from adopting an active conformation. As the structure of each switch pocket is relatively unique for a particular kinase, and must be conserved to retain function, drugs acting at this region can be targeted to specific kinases and are theoretically less likely to be affected by resistance mutations. Three compounds (DP-2976, DP-3636 and DP-4444) are now in early preclinical development and have demonstrated superior in vitro potency against resistant GIST cell lines compared with imatinib and sunitinib [Heinrich et al. 2010].
Targeting downstream signaling and primary mutations
Inhibition of downstream targets of receptor tyrosine kinases is also under investigation and may be complementary to ongoing direct KIT/PDGFRA inhibition. PI3K/AKT/mammalian target of rapamycin (mTOR) signaling appears particularly important in imatinib-resistant GIST [Bauer et al. 2007]. A number of PI3K inhibitors are now in early clinical development, with the Novartis compound BKM120 planned for a phase I study in GIST in early 2012, and other compounds (e.g. BEZ235, XL147, GDC-0941) being investigated in patients with advanced solid tumors. The mTOR inhibitor everolimus, in combination with imatinib, has completed a phase II study in 47 patients with GIST refractory to imatinib and sunitinib with a reported disease control rate of 45%, meeting its predefined efficacy criteria [Schoffski et al. 2010]. Dual PI3K/mTOR inhibitors such as PI-103, NVP-BEZ235, XL675 and GDC-0980 have also demonstrated promising preclinical activity in a range of tumor types, with the latter two being tested as monotherapy in phase I clinical trials [Park et al. 2008; Maira et al. 2008; Serra et al. 2008]. Early reports are supportive of meaningful clinical activity, with a single patient with GIST treated with GDC-0980 obtaining a robust metabolic response based on FDG-PET and continuing on the study for greater than 6 months [Wagner et al. 2011].
The RAS/RAF/MEK pathway is another attractive target in GIST, especially in the small number of KIT/PDGFRA wild-type tumors that harbor BRAF V600E mutations [Agaram et al. 2008; Agaimy et al. 2009]. Compounds that target this pathway, including selective BRAF inhibitors and MEK inhibitors, have demonstrated dramatic clinical activity in BRAF V600E mutant melanoma; however, they have been less effective in other tested BRAF mutant solid tumors, and are as yet untested in patients with GIST [Chapman et al. 2011; Infante et al. 2011]. Selecting therapies based on primary oncogenic drivers is also being furthered by two new PDGFR-specific drugs: the tyrosine kinase inhibitor, crenolanib (CP 868-956), and a monoclonal antibody, olaratumab (IMG-3G3). These compounds are active against the common, imatinib- and sunitinib-resistant PDGFRA exon 18 D842V mutation – with clinical trials underway focusing on this patient population [Lewis et al. 2009; Shah et al. 2010]. Finally, in patients with wild-type GIST, new anti-IGF-1R monoclonal antibodies and small molecule tyrosine kinase inhibitors may be of selected benefit. Compounds currently in development have demonstrated single agent activity in some sarcoma subtypes, and these drugs may be effective in the subgroup of patients whose tumors have amplification or overexpression of IGF-1R [Day et al. 2010].
Closing thoughts
Within the last decade our improved knowledge of the oncogenic drivers and resistance mechanism operant in GIST have been dramatic, and has acted as a foundation for our general understanding of the role of targeted therapies in human cancers. Despite this, our overall approach to the management of GIST remains largely empirical. Imatinib remains the only approved first-line therapy, and whilst its effectiveness for the majority of patients has been revolutionary, a well defined subset can be anticipated not to do as well. For now, first-line management strategies for patients with advanced GIST remain focused on optimizing response to initial imatinib therapy. Sunitinib as the only approved second-line agent will be effective for some, but again, many nonresponders can be fairly accurately predicted in advance. Personalizing the treatment of GIST is a challenge for the next decade. Tailoring treatments to tumor genotype, and broad-activity or combination therapies to prevent emergence of resistance, will be essential to optimize patient outcomes. The ongoing support of well designed clinical trials, testing promising new compounds – some hopefully described in this review – will be required in order to get there.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
JZ: Novartis – honoraria for advisory boards, travel support, research support; Pfizer – honoraria for speaker and grant panel support, research support; Bayer – research support. DK: Bristol Myer-Squibb – honoraria speaker, travel support.