
Abstract
Background:
Lymphocyte-activation gene 3 (LAG-3), a checkpoint molecule contributing to immune suppressive microenvironment, is regarded as a promising target in cancer treatment. SHR-1802 is a novel anti-LAG-3 monoclonal antibody.
Objectives:
To evaluate the safety, tolerability, pharmacokinetics, and antitumor activity of SHR-1802.
Design:
A phase I dose-escalation and expansion trial of SHR-1802 in patients with advanced solid tumors.
Methods:
Patients with confirmed advanced solid tumors who failed previous standard-of-care or for whom no effective therapy was available were enrolled to receive SHR-1802 once every 21-day cycle. Dose escalation was performed in an accelerated titration design followed by a 3 + 3 scheme at escalating doses from 0.3 to 10 mg/kg. On the basis of results from dose-escalation phase, one or two dose levels were expanded to establish the recommended phase II dose (RP2D). The primary end points were dose-limiting toxicity (DLT) and RP2D.
Results:
Between 01 July 2020, and 07 September 2021, 28 patients were enrolled. No DLTs were observed, and all doses investigated were well tolerated. Treatment-related adverse events occurred in 20 patients (71.4%), all grade 1 or 2, with the most common ones being anemia (14.3%), asthenia (14.3%), electrocardiogram QT prolonged (14.3%), followed by increased blood fibrinogen (10.7%), infusion-related reaction (10.7%), and hypoalbuminemia (10.7%). No adverse event-related discontinuation occurred. Three patients died from adverse events, but none of the deaths were deemed related to study treatment. SHR-1802 exposure enhanced with the increasing doses in a greater than dose-proportional manner over the investigated dose range. The disease control rate was 32.0% (95% CI 14.9%–53.5%). The median progression-free survival was 2.0 months (95% CI 1.2–6.1).
Conclusions:
SHR-1802 demonstrated a tolerable safety profile and preliminary antitumor activity in patients with advanced solid tumors. Further studies with larger sample size and in combination forms are warranted for future clinical application.
Registration ClinicalTrials.gov:
NCT04414150
Introduction
Immune checkpoint inhibitors, including antibodies targeting programmed death 1 (PD-1), programmed death ligand 1 (PD-L1), and cytotoxic T-lymphocyte antigen 4 (CTLA-4), have remarkably revolutionized the treatment landscape of cancer, including melanoma, non-small-cell lung cancer, and renal cell carcinoma.1–5 However, durable clinical outcomes are limited to a subset of patients and many initial responders eventually develop progressive disease, suggesting at least in part the existence of alternative immunosuppressive pathways.
Mainly expressed on activated CD4+, CD8+ T cells, CD4+ regulatory T cells, and natural killer cells,6,7 lymphocyte-activation gene 3 (LAG-3) emerges as a negative immune checkpoint molecule by inducing T-cell exhaustion and impairing T-cell proliferation, making it a potential therapeutic target for cancer treatment.8,9 Increased LAG-3 expression on tumor infiltrating lymphocytes was found in various tumor types, such as hepatocellular carcinoma, Hodgkin lymphoma, ovarian cancer, and melanoma, and inhibiting the interaction of LAG-3 to its ligand can activate T cells and trigger antitumor immunity.9–12 Co-expression of LAG-3 and PD-1 has been reported in previous studies, and LAG-3 exhibits a striking synergy effect with PD-1 to restrict immune responses, thus combinatorial blockade of LAG-3 and PD-1 is believed to exert enhanced antitumor immunity and stronger tumor suppression. 13 RELATIVITY-047 study demonstrated that nivolumab, an anti-PD-1 antibody, plus relatlimab, an anti-LAG-3 antibody, significantly improved progression-free survival (PFS) compared to nivolumab alone in patients with previously untreated metastatic or unresectable melanoma. 14 Based on this result, this combination regimen has been approved by the U.S. Food and Drug Administration for the treatment of patients with unresectable or metastatic melanoma. 15
SHR-1802 is a novel humanized anti-LAG-3 antibody that specifically binds to LAG-3 and inhibits ligand binding by major histocompatibility complex class II (MHC-II), fibrinogen-like protein 1 (FGL1), galectin-3, and liver sinusoidal endothelial cell lectin.12,16,17 Preclinical studies show that SHR-1802 binds to human LAG-3 protein and Chinese hamster ovary suspension cells expressing human LAG-3, with a half-maximal effective concentration of 0.013 and 0.017 µg/mL, respectively. Preclinical models also show an acceptable safety profile, with a maximum tolerated dose (MTD) of 30 mg/kg in mice and 200 mg/kg in cynomolgus monkey. SHR-1802 combined with camrelizumab, an anti-PD-1 antibody, showed superior tumor suppression in mice compared to single agent with either SHR-1802 or camrelizumab (unpublished data). Based on the aforementioned data, we conducted this phase I dose-escalation and expansion study, aiming to assess the safety and tolerability, establish the MTD and recommended phase II dose (RP2D), as well as evaluate pharmacokinetics and preliminary antitumor activity of SHR-1802 in patients with advanced solid tumors.
Methods
Study design and patients
In this open-label, phase I study (NCT04414150), patients aged 18 to 75 years with clinically or pathologically confirmed advanced solid tumors who failed previous standard-of-care or for whom no effective therapy was available were enrolled. Patients were required to have at least one measurable lesion according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. Other inclusion criteria included an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1; a life expectancy of at least 12 weeks; adequate bone marrow reserve (absolute neutrophil count ⩾1.5 × 109/L, platelet count ⩾90 × 109/L, and hemoglobin ⩾9 g/dL; treatment with blood products or cell growth factors within 14 days before study drug initiation was not allowed); adequate hepatic function [serum albumin ⩾3.0 g/dL, total bilirubin ⩽1.5 times upper limit of normal (ULN), alanine aminotransferase and aspartate aminotransferase ⩽3.0 times ULN]; adequate renal function (serum creatinine ⩽1.25 times ULN); and a normal cardiac function (left ventricular ejection fraction ⩾50%). The major exclusion criteria included known or suspected autoimmune disease; use of systemic corticosteroids or other immunosuppressive agents within 28 days before study drug administration; severe allergic reactions to other monoclonal antibodies; and previous LAG-3 antibody treatment. Treated brain metastases were permitted if the disease was stable for at least 1 month and steroids treatment (prednisone or equivalent) was discontinued for at least 2 weeks. All patients (except those receiving single lead-in dose) were required to provide formalin-fixed, paraffin-embedded tumor samples.
Treatment and assessments
To determine the starting dose for the dose-escalation phase, one to two patients per dose level were enrolled to receive a single lead-in dose (1 µg/kg or 30 µg/kg intravenously). The 30 µg/kg dose was initiated only if 1 µg/kg showed manageable tolerability during the 21-day cycle. The starting dose of the dose-escalation phase was chosen based on the preclinical testing, results from single dose lead-in phase, and study data of similar drugs. In the dose-escalation phase, an accelerated titration design was introduced for the first dose level followed by a 3 + 3 scheme. The dose levels tested were 0.3, 1, 3, and 10 mg/kg administered intravenously once every 21-day treatment cycle. One patient received the starting dose of 0.3 mg/kg, if dose-limiting toxicity (DLT) or at least two grade 2 or worse treatment-related adverse events (TRAEs) were observed during the first treatment cycle, the 3 + 3 scheme was triggered starting from the 0.3 mg/kg dose; if none, the 3 + 3 scheme was implemented starting from the 1 mg/kg dose. Intra-patient dose escalation was not permitted. The decision to proceed to the next dose level was made based on the safety, tolerability, and pharmacokinetics of the first treatment cycle. The MTD was defined as the highest dose level where less than two of six patients experienced DLTs during the first treatment cycle. After a thorough review of the total data during dose-escalation phase, one or two dose levels were chosen to proceed to the dose-expansion phase for establishment of RP2D. Treatment continued until disease progression, intolerable toxicity, patient withdrawal, or at investigators’ discretion.
Dose interruption out of the DLT observation window was allowed for up to 12 weeks. Doses were discontinued for patients requiring a dose interruption of more than 12 weeks. Dose reduction of SHR-1802 was not permitted.
Safety was assessed following the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI-CTCAE) version 5.0. Adverse events were monitored throughout the study treatment until 90 days after the last dose. Safety follow-up continued until all adverse events resolved to baseline level or grade ⩽1. Other safety assessment, including laboratory tests, vital sign, physical examination, and 12-lead electrocardiography were done on a regular basis.
For single-dose pharmacokinetic evaluation, blood samples of all patients were collected 30 min before dosing, 5 min, 2, 8, 24, 72 h, 7 days, and 14 days after the end of infusion on day 1 of cycle 1. For multiple-dose pharmacokinetic analysis, blood samples of all patients in the dose-escalation and expansion phase were obtained 30 min before dosing and 5 min after the end of infusion on day 1 of each cycle from cycles 2 to 7. From cycle 8, blood samples were only collected 30 min before dosing on day 1 of every 3 cycles.
Tumor response was evaluated following RECIST version 1.1 by CT or MRI at baseline, the first day of cycle 4, and every 3 cycles thereafter until disease progression, start of new anticancer treatment, or withdrawal of consent. Patients who were assessed as complete response or partial response needed a confirmation at least 4 weeks later.
For immunogenicity analysis, blood samples were collected 30 min before dosing, 7 days, and 14 days after the end of infusion on day 1 of cycle 1, 30 min before dosing on day 1 of each cycle from cycle 2 to cycle 7, and 30 min before dosing on day 1 of every 3 cycles thereafter. Blood samples were collected on the day of treatment discontinuation and 30, 60, and 90 days after last dose.
LAG-3 expression, assessed by immunohistochemical staining on tumor tissues, was recorded. LAG-3 positivity was defined as LAG-3 expression in ⩾1% of all immune cells.
Definition of DLTs
DLTs was defined as TRAEs observed during the first cycle of treatment: grade 4 hematologic toxicities (except for decreased lymphocyte count), grade 3 neutropenia accompanied by fever (⩾38.5°C), or grade 3 thrombocytopenia with bleeding; grade 3 non-hematologic toxicities (rash, diarrhea, nausea, and vomiting lasting for ⩾2 days despite adequate symptomatic treatment), excluding asymptomatic laboratory abnormalities; grade 3 laboratory abnormalities requiring hospitalization or lasting ⩾7 days; toxicities causing dose delay ⩾7 days in the second cycle.
End points
The primary objectives were DLT and RP2D. The secondary end points were safety (assessed by adverse events, laboratory test, vital sign, and frequency of dose interruption and discontinuation due to TRAEs), antitumor activity [including objective response rate (ORR), which was defined as the proportion of patients with confirmed complete response and partial response; duration of response (DoR), which was defined as time from response to disease progression per RECIST version 1.1 or death, whichever occurred earlier; disease control rate (DCR), defined as the proportion of patients with confirmed complete response, partial response, or stable disease; and PFS, defined as time from first dose to first documented disease progression per RECIST version 1.1 or death, whichever occurred earlier], pharmacokinetics [including maximum plasma concentration (Cmax), time to Cmax (Tmax), area under the plasma-concentration curve from time zero to infinity (AUC0-∞), area under the plasma-concentration curve from time zero to time of last measurable concentration (AUC0-t), clearance (CL), elimination half-life (t1/2), mean residence time (MRT), steady-state volume of distribution (Vss), minimum plasma concentration at steady state (Cmin), and accumulation ratio (Rac)], and immunogenicity of SHR-1802 [anti-drug antibody (ADA) incidence included treatment-induced ADA (baseline negative and post-treatment positive results) and treatment-boosted ADA (both baseline and post-treatment positive results where the increase in titer from baseline was ⩾4-fold)]. The exploratory objective was to investigate the association of LAG-3 expression level with treatment efficacy.
Statistical analyses
Approximately two to four patients were required at the single dose lead-in phase. Sample size for dose-escalation phase was determined by accelerated titration design and 3 + 3 scheme rules, and approximately 10–24 patients were needed. For dose-expansion part, we anticipated that 10–24 patients would be required to establish the RP2D with no formal hypothesis testing.
Safety analyses were conducted in patients who received at least one dose of study treatment and had post-dose safety assessments; pharmacokinetic analyses were performed in patients who received at least one dose of study treatment and had at least one measurable plasma concentration (concentration analysis) or pharmacokinetic parameter (parameter analysis); efficacy analysis set included all patients who received at least one dose of study treatment. Immunogenicity analysis population included all patients who received at least one dose of study drug, had baseline, and at least one post-baseline ADA assessment. Safety and pharmacokinetics were summarized descriptively. ORR and DCR were calculated, and their 95% confidence intervals (CIs) were estimated with Clopper–Pearson method. Kaplan–Meier methods was introduced to estimate median PFS and DoR, and their corresponding 95% CIs were calculated with Brookmeyer–Crowley method on the basis of a log–log transformation. Pharmacokinetic parameters were calculated with standard non-compartmental methods using Phoenix WinNonlin version 8.1.0. All statistical analyses were done with SAS version 9.4.
Results
Patient baseline characteristics and disposition
Between 01 July 2020 and 07 September 2021, a total of 28 patients were enrolled, with three patients receiving single lead-in doses (1 µg/kg dose n = 1; 30 µg/kg dose n = 2), 11 patients in the dose-escalation phase (0.3 mg/kg dose, n = 1; 1 mg/kg dose, n = 3; 3 mg/kg dose, n = 4; 10 mg/kg dose, n = 3), and additional 14 patients in the dose-expansion phase (3 mg/kg dose, n = 7; 10 mg/kg dose, n = 7). As of cutoff date on 13 January 2022, the median follow-up duration was 3.0 months (range 0.7–6.7). In all, 25 patients discontinued study treatment, and the major reason was radiographic progression (15/25, 60.0%; Figure 1). Patients enrolled had a wide variety of solid tumors, including lung cancer, esophageal carcinoma, rectal cancer, and nasopharyngeal carcinoma, etc. (Table 1). All patients had a ECOG performance status of 1. All patients had metastasis, with 16 patients (57.1%) carrying three or more metastatic sites.

Trial profile.
Patient characteristics at baseline.
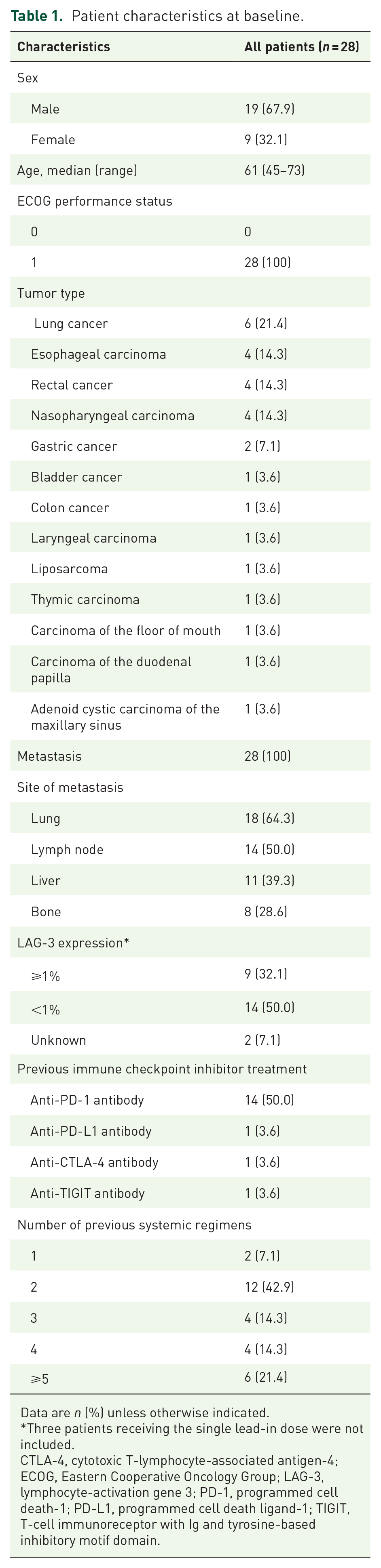
Data are n (%) unless otherwise indicated.
Three patients receiving the single lead-in dose were not included.
CTLA-4, cytotoxic T-lymphocyte-associated antigen-4; ECOG, Eastern Cooperative Oncology Group; LAG-3, lymphocyte-activation gene 3; PD-1, programmed cell death-1; PD-L1, programmed cell death ligand-1; TIGIT, T-cell immunoreceptor with Ig and tyrosine-based inhibitory motif domain.
Safety
All patients were included in the safety analysis set. The median number of treatment cycles was 3.0 (range 1–10). No DLTs were observed in the dose-escalation phase, and all doses studied were well tolerated. Treatment-emergent adverse events (TEAEs) were reported in 26 patients (92.9%). Among them, seven patients (25.0%) had grade ⩾3 TEAEs. TRAEs occurred in 20 patients (71.4%), and all were grade 1 or 2. Five patients (17.9%) experienced serious adverse events, with two patients (18.2%) in the 3 mg/kg cohort and three (30.0%) in the 10 mg/kg cohort (disease progression n = 3; cholecystitis, appendicitis, and abdominal pain upper n = 1 each). None of the serious adverse events were deemed to be related to study treatment. The most commonly reported TRAEs were anemia (14.3%), asthenia (14.3%), electrocardiogram QT prolonged (14.3%), increased blood fibrinogen (10.7%), infusion-related reaction (10.7%), and hypoalbuminemia (10.7%; Table 2). No TEAEs-related dose interruption or discontinuation occurred. Immune-mediated adverse events were observed in seven patients (25.0%). Three patients (10.3%), two in 3 mg/kg cohort and one in 10 mg/kg group, died due to TEAEs (all as a result of disease progression), and none were deemed to be treatment related.
Treatment-related adverse events.

Data are n (%).
Pharmacokinetics
Pharmacokinetic data were available for doses ranging from 30 µg/kg to 10 mg/kg. Mean concentration–time curves after single and multiple administration of SHR-1802 among the ascending doses are presented in Figure 2. The lower limit of quantitation for SHR-1802 was 0.0125 µg/mL. The pharmacokinetic parameters are summarized in Table 3. After single dosing, Tmax was reached shortly after the infusion, and the median results were mainly between 0.67 and 1.10 h, with an exception in 1 mg/kg cohort (2.90 h). However, the range of Tmax was similar between the 1 and 10 mg/kg cohort, and the longer median Tmax in the 1 mg/kg cohort might be attributed to the distinct individual difference and the small sample size. SHR-1802 exposure (Cmax, AUC0-t, and AUC0-∞) increased with the ascending doses ranged from 30 µg/kg to 10 mg/kg after single administration. Dose proportionality assessment of SHR-1802 over a range of 30 µg/kg to 10 mg/kg suggested that SHR-1802 exposure increased greater than a dose-proportional manner over this dose range (Table 4). t1/2 and MRT showed a trend toward dose-dependent increase. Vss appeared similar among the dose range investigated, and no apparent dose-dependent trend was observed. CL decreased substantially from 30 µg/kg to 3 mg/kg dose level and appeared consistent at doses of 3 mg/kg and 10 mg/kg. After multiple infusion, geometric mean Cmax,ss was 53.6 µg/mL in the 3 mg/kg cohort and 208 µg/mL in the 10 mg/kg cohort, and Rac for the Cmax in the two cohorts were 1.18 and 1.14, respectively. Rac for Cmin was 1.80 in 3 mg/kg cohort and 1.92 in 10 mg/kg cohort, indicating mild accumulation after repeated infusion. In the 0.3 mg/kg cohort, there seemed to be a higher Rac for Cmax (3.87); however, given that only one patient in this cohort, no definitive conclusion could be drawn.

Mean plasma concentration (±standard deviation) after single dose ((a) linear plot; (b) semi-log plot) and multiple doses ((c) linear plot; (d) semi-plot) of SHR-1802.
Plasma pharmacokinetic parameters of SHR-1802 following single and multiple doses.

AUC0-t, area under the plasma-concentration curve from time zero to time of last measurable concentration; AUC0-∞, area under the plasma-concentration curve from time zero to infinity; Cmax, maximum plasma concentration after single dose; Cmax,ss, maximum plasma concentration at steady state; Cmin,ss, minimum plasma concentration at steady state; CL, clearance; CV, coefficient of variance; GeoMean, geometric mean; MRT, mean residence time; Rac1, accumulation ratio of Cmax; Rac2, accumulation ratio of Cmin; SD, standard deviation; Tmax, time to Cmax; t1/2, elimination half-life; Vss, steady-state volume of distribution.
Dose proportionality assessment for SHR-1802.

AUC0-t, area under the plasma-concentration curve from time zero to time of last measurable concentration; AUC0-∞, area under the plasma-concentration curve from time zero to infinity; Cmax, maximum plasma concentration after single dose.
Efficacy
All 25 patients in the dose-escalation and dose-expansion phase were included in the efficacy analysis set. No patients had complete or partial response. Eight patients had stable disease, with five patients in the 3 mg/kg group and three in the 10 mg/kg group, and the DCR was 32.0% (95% CI 14.9%–53.5%). The duration of stable disease ranged from 63 to 187 days. The best changes in tumor size from baseline are present in Figure 3. The median PFS was 2.0 months (95% CI 1.2–6.1). LAG-3 expression was available in 23 of all 25 patients. Nine patients (39.1%) demonstrated positive LAG-3 staining, of which three patients had a best response of stable disease, four had progressive disease, and two were not assessable.

The best percentage change from baseline in target lesions. Patient numbers 1, 8, 10, 11, 17, and 20 had lung cancer; patient numbers 2, 5, 6, and 12 had esophageal carcinoma; patient numbers 3, 4, and 14 had rectal cancer; patient number 7 had gastric cancer; patient number 9 had carcinoma of the duodenal papilla; patient number 13 had liposarcoma; patient number 15 had thymic carcinoma; patient numbers 16, 19, and 22 had nasopharyngeal carcinoma; patient number 18 had carcinoma of the floor of mouth; and patient number 21 had adenoid cystic carcinoma of the maxillary sinus.
Immunogenicity
Anti-SHR-1802 antibody was detected in three patients (10.7%), with one patient in the 0.3 mg/kg cohort and two in the 1 mg/kg cohort. All ADA responses were treatment induced. The median time from treatment initiation to first detection of ADA was 43 days (range 22–63).
Discussion
In this phase I, dose-escalation and dose-expansion trial, we assessed the safety, tolerability, pharmacokinetics, and preliminary antitumor activity of SHR-1802 in patients with advanced malignancies. SHR-1802 exhibited well tolerability in different doses. The MTD was not reached at doses up to 10 mg/kg. Dose escalation of SHR-1802 beyond 10 mg/kg level was not conducted as the CL decreased substantially along with doses increasing from 30 µg/kg to 3 mg/kg, displaying a nonlinear elimination owning to target-mediated drug disposition; following doses of 3 mg/kg and 10 mg/kg, SHR-1802 showed linear elimination kinetics, suggesting that the receptors may be fully saturated with SHR-1802. Thus, despite the lack of MTD, 3 mg/kg and 10 mg/kg doses were selected for further exploration on the basis of pharmacokinetic parameters. Studies evaluating safety and efficacy of SHR-1802 at doses of 3 mg/kg and 10 mg/kg in combination with other immune checkpoint inhibitors are being conducted to establish the recommended dose.
SHR-1802 was well tolerated. TRAEs occurred in 71.4% of patients with no grade ⩾3 TRAEs reported. 17.9% of patients experienced serious adverse events, but none were deemed related to study treatment. No patients discontinued study treatment because of adverse events. Anemia, asthenia, abnormal laboratory tests, infusion-related reaction, and gastrointestinal toxicities were the most common TRAEs. The spectrum and incidence of TRAEs of SHR-1802 were consistent with that reported for favezelimab, another anti-LAG-3 antibody. 18 However, in the study investigating favezelimab monotherapy in patients with advanced microsatellite stable colorectal cancer, TRAEs of grade 3 or 4 were observed in 15.0% of the patients, while in our present study, all TRAEs were grade 1 or 2. 18
Following infusion, SHR-1802 exposure (Cmax, AUC0-t, and AUC0-∞) increased with the ascending doses in a greater than dose-proportional manner over the dose range of 30 µg/kg to 10 mg/kg. Multiple infusions of SHR-1802 induced mild accumulation for Cmax. SHR-1802 exposure displayed a trend comparable to the pharmacokinetic profile of ieramilimab. 19
Preliminary antitumor activity was seen with SHR-1802. Although no confirmed complete or partial response was identified in this study, stable disease was observed in eight patients (32.0%), and five of which showed tumor size reduction from baseline. Similar findings were observed in other study, in which limited antitumor activity was observed with LAG-3 inhibitor monotherapy. 19 It has been reported that combination of LAG-3 inhibitors with PD-1 or PD-L1 inhibitors can exert better antitumor efficacy.14,19 In a phase I/II study for patients with advanced malignancies, 32 patients (23.9%) had stable disease with ieramilimab (a LAG-3 inhibitor) alone, and no objective responses were reported; while ieramilimab plus apartalizumab (a PD-1 inhibitor) induced 13 complete or partial responses (10.7%) and 35 stable diseases (28.9%). 19 Moreover, in the RELATIVITY-047 trial, combination form of relatlimab and nivolumab demonstrated a significantly longer median PFS than nivolumab alone in untreated advanced melanoma. 14 Inspired by these aforementioned evidence, future studies are warranted to investigate the clinical benefit of SHR-1802 in combination with a PD-1/PD-L1 inhibitor.
Investigation of possible clinical biomarkers indicating treatment benefits would facilitate the advantageous population selection. In a phase I/II study with heavily pretreated melanoma patients, relatlimab plus nivolumab showed an ORR of at least 3.5-fold higher in patients with a LAG-3 expression ⩾1% than those with a LAG-3 expression <1%. However, in the present trial, no obvious association between LAG-3 expression and clinical efficacy was observed. Given the small sample size in our study, whether LAG-3 would work as a predictive biomarker remains to be further determined.
Conclusions
This phase I dose-escalation and dose-expansion study demonstrated that SHR-1802 had a tolerable safety profile. SHR-1802 exposure (Cmax, AUC0-t, and AUC0-∞) increased with the ascending doses investigated in a trend greater than dose proportionality. Preliminary clinical benefit was observed in patients with advanced solid tumors, and additional studies are warranted to further explore the safety and clinical efficacy of SHR-1802, especially in combination form with a PD-1/PD-L1 inhibitor.