
Abstract
[177Lu]Lu-PSMA has recently been approved for use in the post-taxane, post-novel hormonal-agent setting in patients with metastatic castration-resistant prostate cancer. As a beta-emitting radioligand targeting prostate-specific membrane antigen (PSMA), it delivers radiation to cells expressing PSMA on their surface. In pivotal clinical trials, patients were selected for this treatment based on positron emission tomography (PET)/CT imaging, requiring PSMA-avid disease with no evidence of discordant disease on 2-[18F]fluoro-2-deoxy-D-glucose PET/CT or contrast CT scan. Despite exhibiting an optimal imaging phenotype, the response for many patients is not durable, and a minority do not respond to [177Lu]Lu-PSMA at all. Disease progression is inevitable even for those who achieve an exceptional initial response. Reasons for both primary and acquired resistance are largely unknown; however, they are likely due to the presence of underlying PSMA-negative disease not identified on imaging, molecular factors conferring radioresistance, and inadequate delivery of lethal radiation, particularly to sites of micrometastatic disease. Biomarkers are urgently needed to optimize patient selection for treatment with [177Lu]Lu-PSMA by identifying those who are most and least likely to respond. Retrospective data support using several prognostic and predictive baseline patient- and disease-related parameters; however, robust prospective data is required before these can be translated into widespread use. Further, early on-treatment clinical parameters (in addition to serial prostate-specific antigen [PSA] levels and conventional restaging imaging) may serve as surrogates for predicting treatment response. With little known about the efficacy of treatments given after [177Lu]Lu-PSMA, optimal treatment sequencing is paramount, and biomarker-driven patient selection will hopefully improve treatment and survival outcomes.
Background
Over the last 20 years, though the incidence of prostate cancer diagnoses has risen, mortality rates of prostate cancer have decreased in first-world countries, likely a product of earlier detection through the implementation of prostate-specific antigen (PSA) screening and therapeutic advancements. 1 Despite this, localized prostate cancer treated with curative intent still carries a significant risk of disease recurrence (29% risk of biochemical recurrence at 10 years), 2 and up to 10% of patients are diagnosed with de novo metastatic disease. 3 Prostate cancer remains the second most frequently diagnosed cancer and the fifth leading cause of cancer-related death amongst men globally. 1 Over the last decade, an increasing armamentarium of treatments has become available to treat metastatic disease, including taxane chemotherapy, novel hormonal agents (NHAs) and poly-[adenosine diphosphate-ribose]-polymerase inhibitors (PARPi). Despite these options, progression to castration resistance remains inevitable, and treatment intent remains palliative. For patients with metastatic castration-resistant prostate cancer (mCRPC), the prognosis remains limited, 4 and disease-related symptoms and complications frequently impact on quality of life.
The advent of targeted prostate-specific membrane antigen (PSMA)-based radioligand therapy heralded a welcome transformation of the advanced prostate cancer treatment landscape and has now been widely adopted into standard practice. [177Lu]Lu-PSMA is a radioactive compound whereby lutetium-177 (177Lu), a beta-emitting radioisotope, is combined with a PSMA ligand and administered intravenously. The radioligand therapy binds to the extracellular domain of PSMA, a transmembrane glycoprotein highly expressed on prostate cancer cells. [177Lu]Lu-PSMA is thereafter internalized via endocytosis, and the PSMA receptor will either undergo lysosomal degradation or be recycled within the cell membrane. The 177Lu delivers high doses of radiation to cells within an average range of 0.7 mm. PSMA is an excellent therapeutic target owing to its relative specificity to prostate cancer cells, particularly in advanced disease where expression is upregulated, with minimal significant physiological uptake on PET/CT of normal organs, such as the salivary glands and kidneys.5–7 Since 2015, mounting evidence has supported the use of [177Lu]Lu-PSMA in mCRPC, culminating in the pivotal single-arm phase II ‘LuPSMA’ trial, which evaluated this treatment in an expanded 50-patient cohort at the Peter MacCallum Cancer Centre in Australia.8,9 Promising high PSA response rates (64% achieved a PSA decline of ⩾50%) led to the phase II randomized TheraP trial, which compared [177Lu]Lu-PSMA-617 to cabazitaxel in men with progressive mCRPC. 10 Hofman et al. found that, compared to cabazitaxel, [177Lu]Lu-PSMA-617 was better tolerated and resulted in improved PSA response rates (66% versus 37% by intention to treat; difference 29% [95% CI 16–42], p < 0.0001) as well as better pain responses. 10 In this trial, [68Ga]Ga-PSMA-11 PET/CT and 2-[18F]fluoro-2-deoxy-D-glucose (FDG) PET/CT imaging were utilized for patient selection. Patients were required to have PSMA-positive disease, defined as SUVmax ⩾20 at one site of disease, with all other measurable sites having SUVmax ⩾10 and no discordant FDG-avid sites of disease. The landmark VISION trial was the first study to demonstrate an overall survival (OS) benefit with [177Lu]Lu-PSMA-617 when combined with protocol-defined best standard care, compared to best standard care alone (median OS 15.3 versus 11.3 months; hazard ratio [HR] 0.62 [95% CI, 0.52–0.74], p = < 0.001). 11 To be eligible for this trial, patients were also required to have PSMA-positive disease; however, this was defined as having greater PSMA uptake than the liver (usually SUVmax 5-7). Although no FDG PET/CT scan was required, patients were ineligible if they had PSMA-negative disease, defined as PSMA uptake equal or less than the liver for their definition of measurable disease. The PSA response rate was 20% lower than the TheraP trial, at 46%, which is in keeping with the less stringent eligibility criteria allowing a more heterogeneous population to be treated. Following this, [177Lu]Lu-PSMA-617 has been approved by the Food and Drug Administration (FDA) and added to the therapeutic arsenal in the post-taxane and post-NHA setting worldwide. Though [177Lu]Lu-PSMA-617 remains the only compound radioligand with regulatory approval for use in mCRPC, other variants exist, such as [177Lu]Lu-PSMA-I&T, which demonstrates similar absorbed doses, toxicity and clinical responses.12,13
Despite a suitable imaging phenotype, however, not all patients respond to [177Lu]Lu-PSMA therapy. In the TheraP trial, patients were only eligible for enrolment if they met the strict PET phenotypic criteria outlined above. Despite this, 17% of enrolled patients showed no response with continued PSA progression, suggesting an underlying primary radio-resistance mechanism at play in a subset of this cohort. Further, for those patients who achieved an initial reduction in PSA, responses are typically not durable. In the TheraP trial the median progression-free survival (PFS) was identical at 5.1 months in each arm, though the hazard ratio favoured [177Lu]Lu-PSMA therapy (HR 0.63 [95% CI 0.46–0.86], p = 0.0028) as the benefit became most apparent after 6 months. 10 Even for patients who exhibit an initial exceptional response with resolution of visible disease on post-treatment imaging, disease progression remains inevitable on long-term follow-up. 9 Mechanisms for the limited response durability and development of acquired resistance remain largely undetermined but are the focus of ongoing research. Though some approaches exist already to monitor treatment response once [177Lu]Lu-PSMA has been initiated (such as serial PSA monitoring and repeat imaging), these measures can only identify early disease progression rather than avoid potentially inappropriate patients commencing treatment in the first place.
Outcomes for patients who received subsequent lines of therapy following [177Lu]Lu-PSMA are also largely unknown. Preliminary data from a retrospective study of 41 patients in Australia suggests that response rates to subsequent treatments after progression on [177Lu]Lu-PSMA are reduced, compared to the expected response rates of each treatment when used earlier in the disease. In this study, the median PSA response rate for all pooled subsequent therapies after [177Lu]Lu-PSMA was 12%, suggesting that optimal sequencing of treatments is essential. 14 In the TheraP trial, all patients at enrolment were suitable candidates to receive either [177Lu]Lu-PSMA-617 or cabazitaxel. Despite this, only 32% of patients treated with [177Lu]Lu-PSMA in the trial went on to receive cabazitaxel on disease progression. 10 Baseline biomarker-driven strategies are urgently needed to optimize patient selection for [177Lu]Lu-PSMA therapy before initiation, to avoid administering potentially ineffective treatment and provide an opportunity for an alternative treatment to be considered. This review will outline the currently available evidence surrounding potentially predictive and prognostic biomarkers associated with [177Lu]Lu-PSMA response and survival outcomes, in addition to discussing potential mechanisms of treatment resistance and how to overcome these (Figure 1).
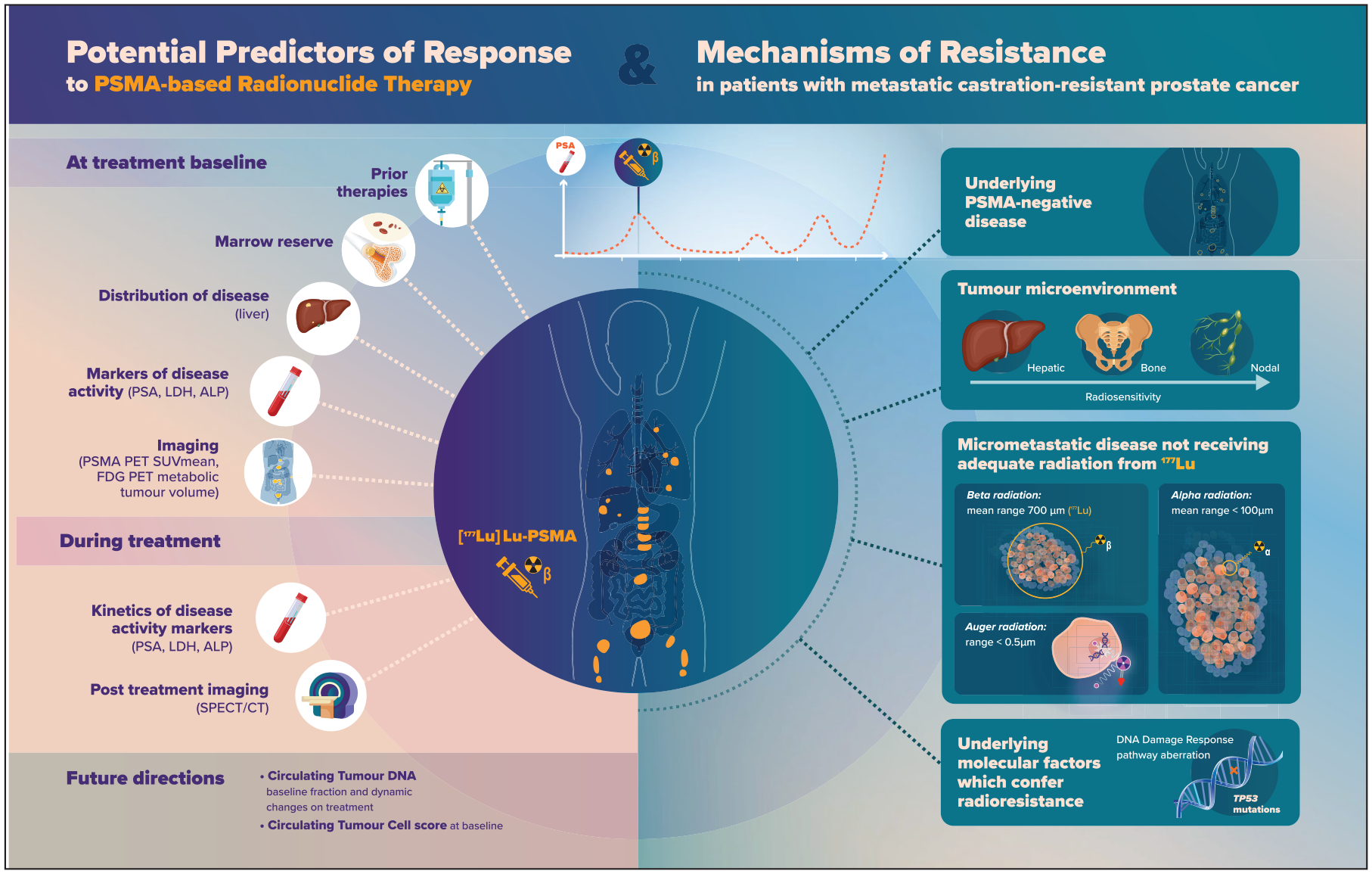
Potential predictors of response and mechanisms of resistance to PSMA-based radionuclide therapy in patients with metastatic castration-resistant prostate cancer.
Baseline predictors of response or resistance to [177Lu]Lu-PSMA
Several analyses have identified baseline patient and disease-related factors that correlate with survival and treatment outcomes following [177Lu]Lu-PSMA. Despite only being evaluated in the post-taxane setting in practice-changing clinical trials and being FDA-approved for the same,10,11 retrospective analyses of patients who received [177Lu]Lu-PSMA on compassionate access programmes allowed interrogation of those who were taxane-naïve versus post docetaxel ± cabazitaxel. Prior taxane chemotherapy (either one or two lines) may be prognostic for worse survival outcomes based on retrospective series.15–17 Barber et al. reported the median OS for taxane-pretreated patients to be 10.7 months, compared with 27.1 months in the taxane-naïve group (p ⩽ 0.001). 16 Kessel et al. observed 109 patients treated with [177Lu]Lu-PSMA and found that second-line chemotherapy with cabazitaxel was associated with worse OS outcomes (6.7 months in post-cabazitaxel patients versus 15.7 months in pre-cabazitaxel patients, p = 0.002). Despite longer OS in the taxane-naïve group, when factoring in that [177Lu]Lu-PSMA was given earlier in the disease course and the natural history of prostate cancer, on multivariate analysis, prior chemotherapy was not a significant prognosticator of OS in both studies. Ahmadzadehfar et al. reported similar findings (median OS for chemotherapy-naïve patients 14.6 months versus post docetaxel 10.9 months versus post docetaxel and cabazitaxel 8.9 months, p = 0.001), which remained statistically significant on multivariate analysis. Therefore, the optimal sequencing of [177Lu]Lu-PSMA remains unknown, with several ongoing trials evaluating its efficacy earlier in the prostate cancer disease course, including in the pre-taxane setting.18,19 Interestingly, prior use of an NHA or radium-223 does not correlate with response or survival outcomes following [177Lu]Lu-PSMA.
Distribution of disease is also informative, with hepatic metastases correlating with worse OS regardless of the degree of PSMA expression seen on PET/CT.15,20,21 In a pre-specified subgroup analysis of the VISION trial, patients with liver metastases had less favourable outcomes with [177Lu]Lu-PSMA in terms of OS compared to those without (patients with liver metastases, HR for OS 0.87 [95% CI 0.53–1.43] versus without liver metastases, HR 0.62 [0.51–0.76]), though this data is limited by a small sample size. This was in contrast to the PFS which favoured patients with liver metastases (patients with liver metastases, HR for PFS 0.28 [95% CI 0.15–0.53] versus without liver metastases, HR 0.43 [0.33–0.57]), suggesting that the poor OS outcomes are related to the inherent poor prognosis of liver metastases rather than a lack of treatment efficacy. Not all sites of visceral disease, however, confer a negative survival outcome, with pulmonary metastases demonstrating favourable responses to [177Lu]Lu-PSMA in several analyses,17,22 suggesting that the underlying tumour microenvironment impacts radiosensitivity. Bone metastases have also been associated with worse survival, with node-only disease presenting the most favourable outlook.23,24
In addition to prior treatments and disease distribution, several blood-based biomarkers can provide helpful information on anticipated treatment response. A higher baseline PSA level has been associated with poorer OS than those with lower starting PSA values (PSA first quartile cut-off, p = 0.007). 25 This effect, however, was not statistically significant in other studies on multivariate analysis. 16 Elevated lactate dehydrogenase (LDH) and alkaline phosphatase (ALP) levels at baseline also correlated with worse survival following treatment with [177Lu]Lu-PSMA in several studies.25,26 Reduced baseline haemoglobin (Hb) is also predictive for poor OS15,16,24, and in one study, a higher Hb level correlated with longer PSA PFS (HR 0.32). 24
Interrogation of novel imaging techniques such as PSMA PET/CT has proven essential to patient selection for [177Lu]Lu-PSMA therapy, having been used as a screening tool in the TheraP and VISION trials.10,11 A prespecified exploratory analysis of the TheraP trial examined whether quantitative PET parameters were associated with response and survival outcomes. 27 Buteau et al. identified that patients with an SUVmean of ⩾10 compared with those with SUVmean < 10 on PSMA PET/CT scan were more likely to achieve a ⩾ 50% PSA response (OR 12.19 versus 2.22, p = 0.039). Conversely, high-volume disease seen on baseline FDG PET/CT (defined as a metabolic tumour volume [MTV] of ⩾200 ml) was prognostic for a lower PSA response rate regardless of the treatment received (OR 0.44, p = 0.035). 27 OS was not reported in this analysis, and whether PSA response rate and PSA PFS can be used as surrogate markers for OS in metastatic prostate cancer remains unclear. Though these predictive and prognostic imaging biomarkers are useful, the methods used to calculate the PSMA SUVmean are complex and time-consuming. They may be challenging to adapt to routine clinical care without artificial intelligence-based measurements.
Gafita et al. developed a prognostic nomogram using a combination of baseline PSMA PET/CT and clinical variables to predict outcomes following treatment with [177Lu]Lu-PSMA. 24 Eighteen variables were initially evaluated retrospectively in 270 patients, and ultimately eight were incorporated into an online prognostic risk calculator. Predictors included time since initial diagnosis of prostate cancer, prior chemotherapy (yes/no), baseline Hb, and several PSMA PET/CT parameters: tumour SUVmean, number of metastases (⩾20 versus <20), the presence of pelvic lymph nodes, bone metastases and liver metastases (yes/no for each). This nomogram is available online to the public; however, it is yet to be prospectively validated or adopted into mainstream practice.
On-treatment predictors of response to [177Lu]Lu-PSMA
Though baseline parameters allow optimal patient selection for [177Lu]Lu-PSMA therapy, emerging predictive on-treatment biomarkers may allow for earlier detection of treatment response or resistance than current methods of assessing tumour response. Though serum PSA level is a widely accepted method of monitoring for response, the Prostate Cancer Working Group 3 (PCWG3) Criteria suggest waiting for 12 weeks before confirming PSA progression in patients who demonstrate a continued rise in PSA. 28 Further, conventional restaging imaging is generally performed after 8–12 weeks or after at least two cycles of [177Lu]Lu-PSMA therapy. Biomarkers that indicate likely response earlier in the treatment course may provide reassurance to continue therapy or justify an early switch to an alternate salvage therapy if response is unlikely.
Irrespective of PCWG3 recommendations, longitudinal monitoring of PSA still plays an essential role in predicting treatment response. Any PSA decline after the first treatment cycle is predictive for longer OS.15,29–31 Whether a ⩾ 50% decline in PSA (as compared to any decrease) after the first cycle or at any stage throughout treatment is significantly associated with survival remains controversial, with several studies producing conflicting results. 32 It is also essential to note that some patients exhibit radiographic progression before a rising PSA. Therefore, PSA is rarely used as the sole reason to base treatment decisions on. The kinetics of LDH and ALP are also prognostic, with a continued steady rise in either parameter after [177Lu]Lu-PSMA initiation associated with shorter OS. 25
In addition to conventional and PET/CT imaging, on-treatment single photon emission computed tomography/CT (SPECT/CT) imaging provides a window into predicted tumour response. Post-treatment SPECT/CT acquired at 24 h was incorporated into trial protocols to enable quantitation of [177Lu]Lu-PSMA retention within the tumour and is considered standard practice in many theranostic centres.8,33 High tumour retention following treatment with [177Lu]Lu-PSMA-617 has previously been associated with an increased chance of a PSA response and a longer PSA PFS.34,35 Post-treatment scintigraphic findings were further evaluated in a retrospective study evaluating 301 patients with a diagnosis of mCRPC who were treated with at least two cycles of [177Lu]Lu-PSMA-I&T. Similar to earlier studies, high-scintigraphic uptake post-treatment was associated with a longer PSA PFS (median PSA PFS 24.9 weeks versus 9.0 weeks; HR 0.3 [95% CI, 0.2–0.5], p ⩽ 0.0001). OS was not significantly different when comparing high versus low tumour uptake on post-treatment SPECT/CT. However, for the subgroup of patients without visceral metastases, the benefit extended to OS for those with high tumour retention (median OS 15.5 months versus 11.4 months; HR 0.6 [95% CI, 0.4-1.0], p = 0.03). Scintigraphic response (defined as > 0.5 cm decrease in infiltration length between the first and second cycle) on serial post-treatment imaging was also prognostic for longer PSA PFS and OS when compared to those who had stable disease or had progressed (median PSA PFS 33.1 versus 16.0 versus 9.0 weeks, p ⩽ 0.0001; median OS 16.5 versus 11.6 versus 7.4 months; p ⩽ 0.0001). 26 These results are in keeping with the known correlation between whole-body tumour radiation dose and PSA response, with the mean radiation dose significantly higher in responders to [177Lu]Lu-PSMA than in non-responders (median 14.1 Gy versus 9.6 Gy). 34 Performing routine dosimetry estimation through post-treatment SPECT/CT imaging is therefore of paramount importance to ensure optimal delivery of and response to [177Lu]Lu-PSMA.
Future directions for biomarker-directed patient selection
In the era of precision medicine where the focus has turned to interrogating the molecular landscape of prostate cancer, several emerging translational markers are under evaluation. Kessel et al. evaluated plasma samples from 19 patients with mCRPC who received [177Lu]Lu-PSMA therapy. 36 All patients had detectable circulating tumour cells (CTCs) at baseline before the first treatment with [177Lu]Lu-PSMA. CTC score correlated with tumour volume measured on PET/CT scan and baseline PSA level; however, it did not correlate with PFS or OS, though small numbers limit this study. Fettke et al. used a targeted next-generation sequencing (NGS) assay to analyse plasma and matched leucocyte samples from a cohort of 13 patients with mCRPC receiving [177Lu]Lu-PSMA. Samples were taken at baseline and before cycle 2. Circulating tumour DNA (ctDNA) was detectable in 89% of the samples, with a median fraction of 26% (range 0–89%). 37 The median ctDNA fraction of patients who experienced a PSA response was 49%, compared to 10% in those who did not respond (p = 0.1). Further, all patients who did not have a reduced ctDNA fraction from baseline to cycle two did not have a PSA response. Limited by small patient and sample numbers, more extensive studies are ongoing to evaluate further the significance of baseline ctDNA fraction and dynamic on-treatment changes.
Mechanisms of resistance to [177Lu]Lu-PSMA therapy
On long-term follow-up of the expanded patient cohort enrolled in the LuPSMA trial, Violet et al. observed that PSA progression is inevitable, even for those who achieved an initial exceptional imaging response. 9 Further, the most common radiographic pattern of progression was osseous, with 56% of patients developing progressive marrow disease. This is despite the study only enrolling patients with ‘PSMA positive’ disease determined by PSMA PET/CT. As aforementioned, a proportion of patients also exhibit primary resistance to treatment despite an optimal imaging phenotype. There is, therefore, an imperative need to dissect the intrinsic and acquired resistance mechanisms to better select patients for [177Lu]Lu-PSMA therapy.
The most likely reason for poor response to [177Lu]Lu-PSMA is the presence of PSMA-negative disease. To this extent, macroscopic PSMA-negative disease can be screened for visually and excluded using PET/CT imaging. In the LuPSMA and TheraP trials, patients underwent screening with both a PSMA and FDG PET/CT and were selected for enrolment based on the results. Patients with PSMA-negative or discordant disease on FDG PET/CT were excluded (16% in LuPSMA, 28% in TheraP).8,10 Similarly, in the VISION trial, patients underwent a PSMA PET/CT (and no FDG PET/CT) and were eligible if they had PSMA-avid disease and no discordant PSMA-negative disease seen on CT scan. 11 Only 12% of patients were excluded, reflecting the less rigorous PET/CT entry criteria. All three trials, therefore, pre-selected patients with an optimal imaging phenotype for treatment with [177Lu]Lu-PSMA. Response rates would be lower in unselected patients, which was demonstrated to an extent when comparing the PSA response rates in the TheraP and VISION trials (66% versus 46%, respectively). It is also clear that patients with a significant burden of FDG-avid disease suffer worse outcomes, suggesting that these patients have a more aggressive disease phenotype. 27 Though the dual PET imaging utilized in the LuPSMA and TheraP trials attempts to exclude most patients with low-PSMA expressing disease, small lesions or micrometastatic disease that cannot be visualized on PET/CT imaging are unable to be evaluated in this manner. Guidelines for patient selection for [177Lu]Lu-PSMA are yet to be established and adopted globally, and practice varies depending on the country and availability of PET imaging. Ideally, patients should have confirmation of PSMA-positive disease with no areas of discordance before treatment, though this is not feasible for many theranostic centres.
Studies are underway to overcome this resistance mechanism by combining [177Lu]Lu-PSMA with a treatment effective against PSMA-negative disease. The LuCAB trial (NCT05340374) is a single-centre study conducted at an Australian site whereby patients receive [177Lu]Lu-PSMA-617 in combination with escalating doses of cabazitaxel. As a taxane, cabazitaxel stabilizes microtubules, causing cell cycle arrest in the most radiosensitive part of the cell cycle (G2-M phase). Cabazitaxel therefore will not just treat underlying PSMA-negative disease but may also enhance the effectiveness of [177Lu]Lu-PSMA through its native radiosensitizing properties. Recruitment commenced in August 2022, and enrolment is ongoing.
Secondly, due to the beta-radiation qualities of 177Lu, micrometastatic disease may not receive adequate radiation to induce cell death. The longer path length of beta-emitters, such as 177Lu (~0.7 mm [range 0.04–1.8 mm]), and lower linear energy transfer (LET), means that a single radiation beam is unlikely to induce cytotoxic double-stranded DNA breaks. Macrotumours rely on crossfire radiation from neighbouring PSMA-positive cells to receive enough cumulative radiation to result in cell death. Areas of micrometastatic disease with single metastatic cells or small cell clusters, such as in the bone marrow, may not receive a lethal radiation dose due to a reduced crossfire effect. To overcome this, several trials are currently underway exploring novel radioisotopes to enhance radiation delivery to micrometastatic disease or combining [177Lu]Lu-PSMA with other therapies. One example is the VIOLET trial (NCT05521412), a phase I/II study evaluating the novel radioligand [161Tb]Tb-PSMA-I&T. Terbium-161 (161Tb), when compared to 177Lu, is theoretically better placed to treat micrometastatic disease. This is due to the higher LET, shorter path length, and presence of Auger and conversion electron emission in addition to the beta emission seen with 177Lu. Auger and conversion electrons have a higher LET rate than beta particles, resulting in greater cell death without reliance on cumulative crossfire radiation. Preclinical data demonstrates that 161Tb delivers higher radiation doses to single tumour cells and micrometastases than 177Lu. 38 Further, in vitro and in vivo results comparing 161Tb to 177Lu demonstrate enhanced therapeutic effects from [161Tb]Tb-PSMA-617, with comparable pharmacokinetics and biodistribution profiles.39–41 Recruitment for the VIOLET trial commenced in October 2022 at the Peter MacCallum Cancer Centre. Similarly, several studies are underway evaluating alpha-emitting radioisotopes in combination with a PSMA-based radioligand (NCT04597411, NCT05219500) or monoclonal antibody (NCT03276572, NCT04506567) in mCRPC. Alpha-emitters have a shorter path length and higher LET, causing more lethal double-strand DNA breaks as they pass through a cell nucleus. In particular, actinium-225 (225Ac) has shown efficacy in mCRPC. Early phase studies have found that combining 225Ac with the PSMA-targeted antibody J591 is efficacious as well as tolerable.42,43 Similarly, retrospective studies have demonstrated clinical activity with [225Ac]Ac-PSMA-617, and it is currently being evaluated in a prospective study (AcTION, NCT04597411). Ongoing trials are also evaluating the combination of [177Lu]Lu-PSMA with an additional radionuclide to cause cumulative radiation, hypothetically resulting in more profound responses. Examples include combining [177Lu]Lu-PSMA-I&T with radium-223 (AlphaBet, 44 NCT05383079) or [225Ac]Ac-J591(NCT04886986).
The underlying tumour microenvironment also appears to impact treatment, with differential responses to [177Lu]Lu-PSMA depending on the metastatic site. This is likely related to the known intra- and inter-tumoral molecular heterogeneity common in advanced disease 45 and varying levels of radiosensitivity depending on the underlying soft tissue origin. As discussed above, hepatic metastases are associated with shorter OS, regardless of PSMA expression. 46 Pulmonary metastases, however, do not confer a negative survival outcome.17,22 Patients with lymph node-only disease have the most favourable outcomes compared to bone and visceral lesions. 47 Bone lesions are a particular area of interest, given that they are the most common site of progression following [177Lu]Lu-PSMA treatment. 9 In addition to marrow disease receiving inadequate radiation, other factors may contribute to the suboptimal response to [177Lu]Lu-PSMA. The bone marrow may contain pro-survival elements not seen in other organs, 48 and bone metastases may also have a higher growth rate when compared to soft tissue metastatic lesions. 49 In general, bone metastases also exhibit lower PSMA expression levels than lymph nodes, which undoubtedly contributes to a less robust response from [177Lu]Lu-PSMA. 50 The aforementioned AlphaBet trial (NCT05383079) is evaluating the combination of [177Lu]Lu-PSMA-I&T with radium-223 (223Ra), with the hope that the bone metastases will receive a higher radiation dose from the bone-specific alpha-emitter 223Ra.
Underlying molecular factors conferring a radioresistant phenotype may be important in the context of primary disease progression. TP53 mediates cell cycle arrest to repair damaged DNA and is a vital effector of the DNA damage response (DDR) pathway. As such, TP53 aberrations have been associated with radioresistance and cancer progression in both murine and human in vitro prostate cancer cell lines.51,52 Other aspects of the DDR pathway have also been implicated in resistance to [177Lu]Lu-PSMA therapy, with some aberrations associated with poor treatment responses. DDR alterations are present in up to 28% of patients with mCRPC. 53 In one study, seven patients who were resistant to treatment with[ 225Ac] Ac-PSMA-617 despite sufficient tumour PSMA expression on PSMA PET/CT underwent a fresh tumour biopsy which was analysed using a targeted DDR-associated gene NGS panel. Kratochwil et al. found a high incidence of DDR aberrations, with the average number of DDR mutations per patient being 2.2 (range 0–6). 54 The PARP enzyme plays a central role in repairing radiotherapy-induced DNA strand breaks, for example, as caused by [177Lu]Lu-PSMA, and minimizes the lethal radiation-induced double-strand DNA damage. 55 It would therefore seem most logical that aberrations in the DDR pathway would confer a radiosensitive advantage. However, this does not appear to be the case with the overrepresentation of DDR alterations seen in Kratchowil’s cohort of radioresistant patients. The DDR pathway is complex, and the effect on radiosensitivity may depend on the specific mutation. Alterations that play a crucial role in DNA damage repair, such as BRCA2,56–59 commonly translate to increased radiosensitivity. Conversely, alterations that affect DNA damage recognition or signalling, such as ATM or CHEK2, promote radioresistance.60,61 The significance of the DDR pathway in this setting remains unclear, however, as several studies show no difference in outcomes in patients with DDR aberrations treated with PSMA-based radioligand therapy when compared to those without a DDR gene defect.62–64 Large-scale prospective translational studies are required to further elucidate the significance of the DDR pathway in responses to [177Lu]Lu-PSMA.
PARPi may produce a radiosensitizing effect by preventing the repair of single-strand and double-strand DNA breaks, thereby promoting cell death. Multiple pre-clinical studies have demonstrated radiosensitizing properties evidenced by an enhanced antitumour effect when a PARPi is combined with radiotherapy. 65 The LuPARP trial (NCT03874884) evaluates the combination of [177Lu]Lu-PSMA-617 with the PARPi olaparib in men with mCRPC, regardless of DDR pathway mutation status. Dose escalation has been completed, and recruitment continues in an expansion cohort.
Conclusion
Integrating [177Lu]Lu-PSMA into the treatment paradigm for mCRPC has been a welcome change in managing a disease that causes significant morbidity and mortality to men globally. However, the benefit of this treatment is limited by the relatively short duration of response for many patients, and it is not effective in all cases. Current practice in many countries includes selecting patients for this therapy based on high PSMA expression, as evidenced by PET/CT imaging. However, this varies depending on local guidelines and available resources. Despite optimal PET/CT imaging, a small proportion of patients demonstrate primary disease progression. Further, little is known about the efficacy of subsequent lines of treatment after [177Lu]Lu-PSMA, highlighting the need to establish its optimal position in the mCRPC treatment sequence. Improved selection of patients for this therapy is an urgent unmet need. As our understanding of the mechanisms of intrinsic and acquired resistance to [177Lu]Lu-PSMA grows, so will our knowledge of valid biomarkers. A combination of baseline patient and disease-related factors and early on-treatment biomarkers may assist with guiding treatment decisions. However, prospective data in large-scale clinical trials are required before such biomarkers can be integrated into clinical practice.