
Abstract
Background:
Aberrant mesenchymal–epithelial transition/hepatocyte growth factor (MET/HGF) regulation presented in a wide variety of human cancers. MET exon 14 skipping, copy number gain (CNG), and kinase domain mutations/arrangements were associated with increased MET activity, and considered to be oncogenic drivers of non-small cell lung cancers (NSCLCs).
Methods:
We retrospectively analyzed 564 patients with MET alterations. MET alterations were classified into structural mutations or small mutations. MET CNG, exon 14 skipping, gain of function (GOF) mutations, and kinase domain rearrangement were defined as actionable mutations.
Results:
Six hundred thirty-two MET mutations were identified including 199 CNG, 117 exon 14 skipping, 12 GOF mutations, and 2 actionable fusions. Higher percentage of MET structural alterations (CNG + fusion) were detected in advanced NSCLC patients. Moreover, MET CNG was enriched while exon 14 skipping was rare in epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKI)-treated advanced NSCLC patients. Ten of the 12 MET GOF mutations were also in EGFR-TKI-treated patients. Fifteen (68.1%) of the 22 patients treated with crizotinib or savolitinib had a partial response. Interestingly, one patient had a great response to savolitinib with a novel MET exon 14 skipping mutation identified after failure of immune-checkpoint inhibitor.
Conclusions:
Half of the MET alterations were actionable mutations. MET CNG, exon 14 skipping and GOF mutations had different distribution in different clinical scenario but all defined a molecular subgroup of NSCLCs for which MET inhibition was active.
Introduction
The hepatocyte growth factor (HGF) receptor, encoded by the mesenchymal–epithelial transition (MET) oncogene, is a receptor tyrosine kinase that drives oncogenesis in many different types of tumors.1,2 Aberrant activation of MET occurs through several mechanisms,3,4 including MET copy number gain (CNG) or rearrangement,4,5 activating point mutations in the kinase domain6,7, MET exon 14 skipping,8–11 which caused increased MET protein levels due to disrupted ubiquitin-mediated degradation of the MET protein. These alterations have been shown to lead to increased MET stability and oncogenic potential, and to confer sensitivity to MET tyrosine kinase inhibitors (TKIs) such as crizotinib, cabozantinib, capmatinib, savolitinib, tepotinib, and many other inhibitors in clinical trials.12–16 Crizotinib is a small molecule multi-kinase inhibitor, which was approved by U.S. Food and Drug Administration for treatment of metastatic ALK-positive and ROS1-positive NSCLC patients in 2011 17 and 2016. 18 MET exon14 skipping and MET amplification mutation NSCLC patients have been reported respond to the crizotinib as well.12,14,19,20 Savolitinib is a highly selective MET-TKI, which has been approved by the China National Medical Products Administration for NSCLC patients with MET exon14 skipping. It also has shown efficacy in NSCLC patients following prior EGFR-TKI treatment with MET amplification.21–23
In the setting of epidermal growth factor receptor (EGFR)–mutant NSCLC, activation of MET has been reported as a primary or secondary resistance mechanism to EGFR TKIs.24–28 This has guided the design of clinical trials testing the activity of EGFR and MET inhibitors in combinations of EGFR-TKI and MET-TKI.29–31 Although it was reported that MET exon 14 skipping happened in approximately 3% of patients with NSCLC,8,9,32,33 it was more enriched in pulmonary sarcomatoid carcinomas.32,34,35 MET was also reported to contribute to cytotoxic chemotherapy resistance. 36 However, there is scant data on the prevalence of other MET activation mutations besides exon 14 skipping in NSCLC, especially for those treatment-naıve or those TKI-treated EGFR-mutant-positive NSCLCs.
In the current study, we aimed to determine the constitution of MET DNA alterations, including nonactionable mutations, actionable CNG, exon 14 skipping, and kinase domain mutations in a large cohort of Chinese patients and to define the characteristics of MET mutation in different stages of tumors. We also presented 22 patients treated with crizotinib or savolitinib including one of them having a novel noncanonical site mutations of MET exon 14 after immunotherapy. Importantly, this heavily treated patient had a durable response to MET-targeted therapy savolitinib.
Methods
Study design
To describe the real-world mutation landscape of MET gene, 564 consecutive NSCLC patients with somatic MET mutations were retrospectively analyzed. Clinical characteristics and treatment histories were annotated, and patients were divided into different groups, according to stages and treatment history. MET mutations were defined as structural mutations (CNG, fusion, and kinase domain duplication) and small mutations [single-nucleotide variants (SNVs) and small insertion deletion, including all MET exon 14 skipping mutations]. Actionable MET mutations were defined as CNG, MET exon 14 skipping mutation, and gain of function (GOF) mutations. Distribution of different types of MET mutation were compared among different patients. Moreover, 22 cases treated with MET-TKI (crizotinib (n = 12) or savolitinib (n = 10)) were presented at the end (Supplemental Figure S1).
DNA extraction, library preparation, and target enrichment
Next-generation sequencing (NGS)-based somatic mutation were performed in a College of American Pathologists-accredited laboratory, Geneplus-Beijing (Beijing, China) from January 2017 to July 2020. All tissue samples included in this study underwent pathology review onsite to confirm histologic classification and the adequacy of the tumor tissues, which required a minimum of 20% of tumor cells. Genomic tumor DNA was extracted from the tumor tissues using the QIAamp DNA Mini Kit (Qiagen, Valencia, CA, USA). Genomic DNA was extracted from white blood cells as a germline control using the DNeasy Blood Kit (Qiagen, Valencia, CA, USA). DNA concentration was measured using a Qubit fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Invitrogen, Carlsbad, CA, USA). Sequencing libraries were prepared from Illumina TruSeq DNA Library Preparation Kits (Illumina, San Diego, CA, USA). Libraries were hybridized to custom-designed biotinylated oligonucleotide probes (NimbleGen SeqCap EZ Library, Roche NimbleGen, Madison, WI, USA), covering ~230 Kbp genomic regions of 59 genes or ~1.4 Mbp genomic regions of 1021 cancer-related genes (Supplemental Tables S1 and S2) using Gene + Seq 2000 instrument.37,38
Sequencing and data analysis
Sequencing data were analyzed using default parameters. Adaptor sequences and low-quality reads were removed. The clean reads were aligned to the reference human genome (hg19) using the Burrows-Wheeler Aligner (version 0.7.12-r1039). GATK (version 3.4-46-gbc02625) was employed for realignment and recalibration. SNVs were called using MuTect (version 1.1.4) and NChot, an in-house software developed for reviewing hotspot variants. Small insertions and deletions (InDels) were determined by GATK. CONTRA (v2.0.8) was used to identify somatic copy number alterations. All final candidate variants were manually verified with Integrative Genomics Viewer. Targeted capture sequencing required a minimal mean effective depth of coverage of 300× and 1000× in tissues and plasma samples, respectively. Targetable genomic alterations simultaneously detected by this assay included base substitutions, short insertions and deletions, focal gene amplifications and homozygous deletions (copy number alterations), and select gene fusions and rearrangements.
Definition of MET alterations
All base substitution, InDel, copy-number alteration, and rearrangement variant reads nearby to the splice junctions of MET exon 14 were examined and then manually inspected to identify those likely to affect splicing of exon 14, or delete the exon entirely. 8 MET CNG was defined as copy number ⩾3. MET fusion was defined if there were more than four split reads and five paired-end reads, and the complete kinase region of the MET gene were retained. 39 MET GOF mutation was defined only if there was reported by literature including cell line studies.
Statistical analyses
All the statistical analyses were performed using GraphPad Prism (v. 8.0; GraphPad Software, La Jolla, CA, USA) software. Associations between any two categorical variables were analyzed with Fisher’s exact test. A two-sided p value of < 0.05 represented statistical significance.
Results
Study design and patient demographics
A total of 564 patients (63.1%, male) of NSCLC with MET mutation or CNG identified in tissue biopsy were analyzed. The median age at diagnosis was 63 (range, 16–86) years, and 298 (52.8%) patients were nonsmokers. There were 474 (84.0%) cases of adenocarcinoma, 68 (12.1%) cases of squamous cell carcinoma, 11 (2.0%) cases of sarcomatoid, 7 (1.2%) cases of adenosquamous carcinoma, and 4 (0.7%) cases of NSCLC other types including large cell carcinoma and mucoepidermoid. Although 107 (19.0%) patients were at the stage of I–IIIa, the rest 457 (81.0%) patients were at the stage of IIIb or IV. For patients at the stage of IIIb or IV, 273 (59.7%) patients were treatment naïve, the rest patients were treated with chemotherapy, targeted therapy, or checkpoint inhibitor previously (Table 1).
Clinicopathological characteristics of patients.
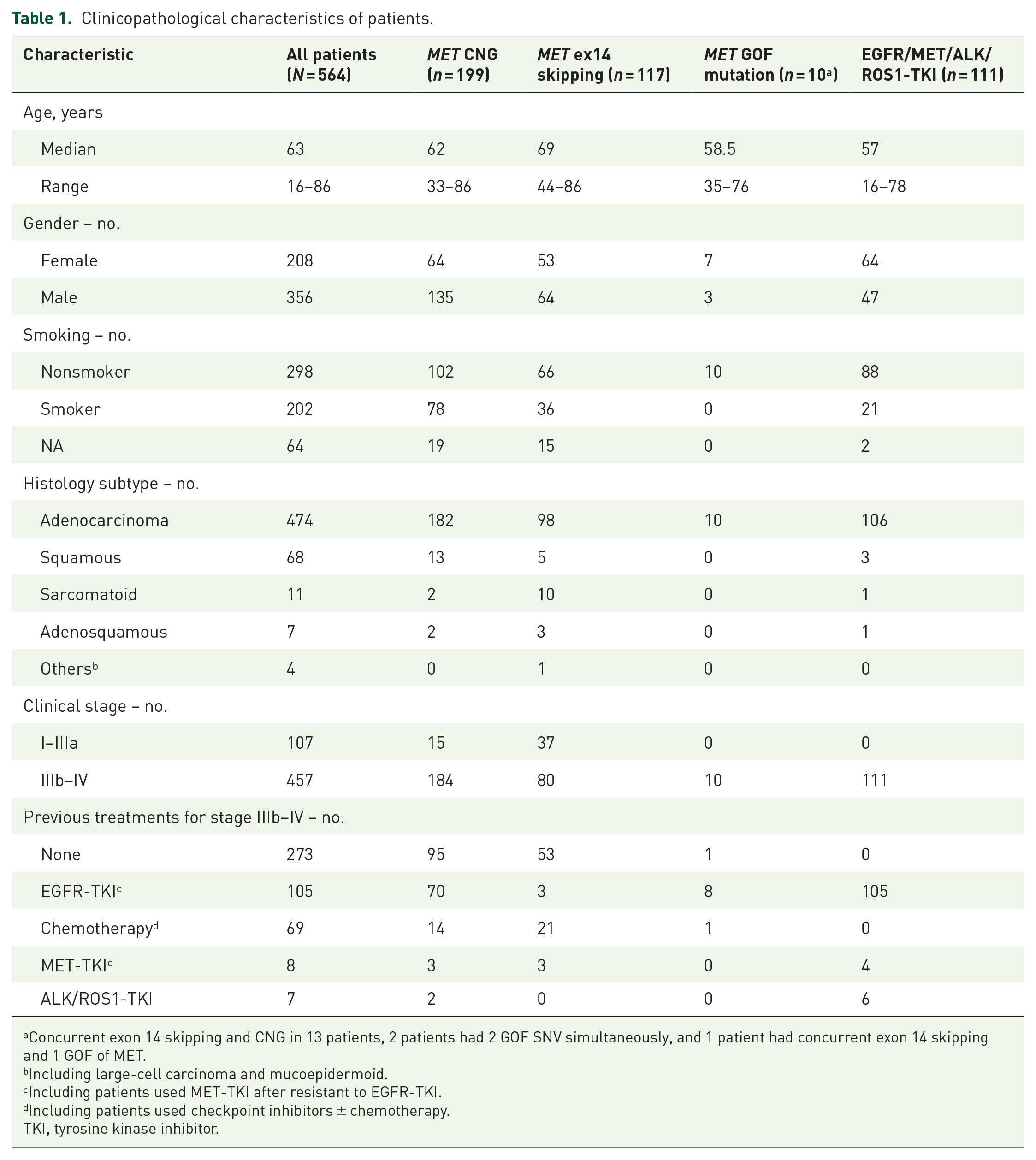
Concurrent exon 14 skipping and CNG in 13 patients, 2 patients had 2 GOF SNV simultaneously, and 1 patient had concurrent exon 14 skipping and 1 GOF of MET.
Including large-cell carcinoma and mucoepidermoid.
Including patients used MET-TKI after resistant to EGFR-TKI.
Including patients used checkpoint inhibitors ± chemotherapy.
TKI, tyrosine kinase inhibitor.
MET mutation profiling
The 642 MET mutations identified in the 564 patients could be divided into large structural variants (CNG + rearrangement) and small mutations (SNV + InDels). Baseline characteristics of the patients by type of MET alteration was presented in Table 1. The percentage of structural variants was significantly lower in stage I–IIIa than stage IIIb–IV patients (13.3% versus 37.5%, p < 0.0001), and it was also significantly lower in treatment-naïve than treated advanced NSCLC patients (32.8% versus 44.4%, p = 0.0074). On the contrary, structural variants was significantly enriched in the EGFR-TKI-treated patients than those treated with chemotherapy, or those treatment-naïve advanced NSCLC patients (63.7% versus 22.2% versus 32.8%, p < 0.0001) (Figure 1(a)–(d)).

Distribution of MET structural mutations (CNG + fusion) versus small mutations (SNV + small Indels), actionable mutations versus nonactionable mutations in different clinical scenario. (a) Number of structural mutation and small mutations in stage I–IIIa and stage IIIb–IV patients with or without systemic therapy. (b) Number of structural mutation and small mutations in stage I–IIIa and stage IIIb–IV patients, p < 0.0001. (c) Number of structural mutation and small mutations in stage IIIb–IV patients with or without systemic therapy, p = 0.0074. (d) Number of structural mutation and small mutations in stage IIIb–IV patients without treatment versus treated with EGFR-TKI versus chemotherapy, p < 0.0001. (e) Overlap between different MET alterations. GOF, gain of function mutation. (f) Number of actionable and nonactionable mutations in stage I–IIIa and stage IIIb–IV patients with or without systemic therapy. (g) Number of actionable and nonactionable mutations in stage I–IIIa and stage IIIb–IV patients, p = 0.040. (h) Number of actionable and nonactionable mutations in stage IIIb–IV patients with or without systemic therapy, p = 0.0055. (i) Number of actionable and nonactionable mutations in stage IIIb–IV patients without treatment versus treated with EGFR-TKI versus chemotherapy, p < 0.001.
Among the 642 MET mutations, 330 (50.9%) were actionable mutations including 199 CNG, 117 exon 14 skipping, 12 GOF SNV, 1 MET fusion, and 1 MET kinase domain duplication. Concurrent exon 14 skipping and CNG were identified in 13 patients, and 2 patients had two GOF SNV simultaneously and 1 patient had concurrent exon 14 skipping and one GOF of MET (Figure 1(e)). The percentage of actionable mutation was lower in stage I–IIIa than stage IIIb–IV patients (43.0% versus 53.4%, p = 0.040), and lower in treatment-naïve than treated advanced NSCLC patients (48.4% versus 60.7%, p = 0.0055). On the contrary, the percentage of actionable mutation was significantly higher in the EGFR-TKI-treated patients than those treated with chemotherapy, or those treatment-naïve advanced patients (71.9% versus 47.6% versus 48.4%, p < 0.001) (Figure 1(f)–(i)).
MET actionable mutations: CNG and fusion
As there was only 15 CNG in stage I–IIIa patients, we focused on stage IIIb–IV for CNG analysis. MET CNG was more common in EGFR-TKI-treated patients than chemotherapy, or treatment-naïve advanced NSCLC patients (66.7% versus 24.3% versus 34.8%, p < 0.01) (Figure 2(a)). The copy number of MET was significantly higher in EGFR-TKI-treated patients than those treatment-naïve advanced patients (6.3 ± 3.8 versus 4.9 ± 2.5, p = 0.0075), but similar with those treated with chemotherapy (6.3 ± 3.8 versus 5.1 ± 3.0, p = 0.25) (Figure 2(b)).

MET copy number gain (CNG) and fusions. (a) Percentage of patients with MET CNG in stage IIIb–IV patients treated with EGFR–TKI versus chemotherapy versus treatment naïve. (b) Copy number of MET CNG in stage IIIb–IV patients treated with EGFR-TKI versus chemotherapy versus treatment naïve. (c) Copy number of EGFR mutant stage IIIb–IV patients treated with or without EGFR-TKI. (d) Allele frequency of MET exon 14 skipping of patients with or without concurrent MET CNG. (e) CD74-MET fusion. (f) MET kinase domain duplication.
MET CNG may occur with other driver mutations: 99 with EGFR mutation, 13 with exon 14 skipping, 8 with KRAS CNG/mutation, 8 with ERBB2 CNG/mutation, 5 with BRAF, 3 with ALK fusion, and 2 with RET fusion. For patients with concurrent EGFR actionable mutation and MET CNG, those treated with EGFR-TKI had significantly higher copy number than those EGFR-TKI-naïve patients (7.3 ± 4.8 versus 4.4 ± 2.4, p = 0.01) (Figure 2(c)). Interestingly, MET CNG occurred with MET exon 14 skipping in 13 patients, we compared the allele frequency of MET exon 14 skipping with or without MET CNG. We found the allele frequency of MET exon 14 skipping was significantly higher in the patients with MET CNG (67.8% ± 19.8% versus 19.8% ± 14.5%, p < 0.0001) (Figure 2(d)). This indicated that if CNG concurred with MET exon 14 skipping, the allele with MET exon 14 skipping was more likely to get CNG.
The CD47-MET fusion was detected in a 46-year-old female lung adenocarcinoma (IV, pleural metastasis), and the MET kinase domain duplication (exon 15–exon 21 duplication) was detected in a 77-year-old male lung adenocarcinoma patient (IV, bone metastasis) (Figure 2(e) and (f)). No other driver mutation but TP53 S240R or C135Y was identified in these two patients.
MET actionable mutations: Exon 14 skipping and GOF missense mutations
In contrast to MET CNG, exon 14 skipping was less common in EGFR-TKI-treated patients compared with chemotherapy, or those treatment-naïve advanced patients (3.7% versus 55.0% versus 35.6%, p < 0.0001). We further analyzed the mutation details of exon 14 skipping. Seventy-five of the 117 cases (64.1%) harbored mutations affecting the splicing donor site, 41 (35.0%) harbored mutations affecting the splicing acceptor site, and 1 case affected the E3 ubiquitin ligase CBL docking site (Y1003). Sixty-three of the 75 cases affecting the splicing donor site had mutations in the canonical splicing site, including 22 cases harbored MET c.3028G>A/T/C mutation, 19 cases harbored c.3028+1G>A/T/C, 11 cases harbored c.3028+2T>A/C/G, and 11 cases harbored insertion/deletion spanning the canonical splicing site. Interestingly, all the 12 cases affecting the noncanonical splicing donor site were c.3028+3A>G/T mutation, including one novel c.3028+3_3028+5delATAinsTTT mutation. On the contrary, all the 41 mutations affecting the splicing acceptor site were deletion with/without insertion, with 15 affecting the canonical site and 26 the noncanonical site (Figure 3(a)). We speculated that the polypyrimidine tract around the splicing acceptor site making it more susceptible to deletion/insertion, and thus SNV less common.

MET exon 14 skipping and gain of function (GOF) mutations. (a) Distribution of MET exon 14 skipping mutations. Pink, exon 14; blue, intron (IVS) 13 or 14; yellow, canonical splicing site; blue bar, noncanonical splicing site deletion/insertion; blue triangle, noncanonical splicing site single-nucleotide mutation. (b) Distribution of MET GOF mutations, all in kinase domain of MET.
Exon 14 skipping co-occurred with EGFR in two patients treated with EGFR-TKI, with KRAS CNG/mutation in two patients, and with ERBB2 CNG in one patient. Though not mutually exclusive to other driver mutations, MET exon 14 skipping concurred less common with other driver mutations than MET CNG.
Notably, for the 10 patients with GOF mutation of MET in the kinase domain, 8 were previously treated with EGFR-TKI, 1 was previously treated with chemotherapy due to the concurrence of MET GOF and MET exon 14 skipping, and 1 was treatment-naïve (Figure 3(b)). This indicated that MET GOF mutation was not a common driver mutation during tumorigenesis, and it may be enriched by EGFR-TKI therapy and acts as one of the resistant mechanisms to EGFR-TKI.
As the patients pretreated with EGFR/ALK/ROS1 TKI may represent a different population with the whole cohort, we summarized the clinical characteristics of this cohort (n = 111) in Table 1 and analyzed the MET alteration and concurrent driver mutations. As expected, the median age of this cohort was significantly younger than other stage IIIb–IV patients (n = 346), and there were more female and nonsmoking patients (p < 0.0001). Moreover, this cohort had higher frequency of MET CNG (72/111 versus 112/346, p < 0.0001), MET GOF mutation (7/111 versus 3/346, p < 0.001), but lower frequency of MET exon 14 skipping (6/111 versus 74/346, p < 0.0001). For the 105 EGFR-TKI-treated patients, 101 patients retained EGFR mutation, with 70 MET CNG, 3 MET exon 14 skipping (including 1 concurrent MET CNG and exon 14 skipping), 9 MET GOF mutations (including two patients with two GOF SNV simultaneously), and 1 NCOA4-RET identified as the resistant mechanism. While all the four ALK-TKI-treated patients retained ALK fusion, with two MET CNG, one ALK G1202R identified as the resistant mechanism.
Efficacy of MET-TKIs in MET actionable mutations
In our cohort, 22 patients were treated with crizotinib (n = 12) or savolitinib (n = 10) as the first (n = 10), second (n = 10), or further-line treatment (n = 2). Among the 22 patients, 6 had MET CNG, 14 had MET exon 14 skipping, and 2 had kinase domain mutation (H1094Y). Moreover, five patients had concurrent EGFR mutation, and three were treated with crizotinib combined with the first-generation EGFR-TKI [Figure 4(a) an-, striped bar, progression-free survival (PFS): 3, 3, and 8 months respectively], whereas two were treated with crizotinb followed EGFR-TKI. Fifteen (68.1%) of the 22 patients had a partial response (PR), 7 had stable disease (SD) (31.9%). The median PFS (mPFS) for all patients was 10 months. The median PFS for patients treated with crizotinib or savolitinib was 8 or 49 months, respectively (hazard ratio [HR] = 5.305, 95% CI 1.764–15.96, p = 0.0342) (Figure 4(a) and (b)). However, patients with MET exon 14 skipping (n = 16) or CNG (n = 4) had comparable PFS in our cohort (mPFS 11.5 versus 8.5 m, HR = 0.4442, 95% CI 0.1028–1.919, p = 0.14). Due to the limited number and retrospective analysis of this MET-TKI cohort, further conclusions should be drawn with cautions.

Response of MET-TKI treatment. (a) Duration of response by crizotinib. (b) Duration of response by savolitinib. (c) Response to crizotinib in the patient (M519) with MET p.H1094Y mutation.
One of the two patients with kinase domain mutation (H1094Y) had a PR with crizotinib as the third-line therapy. This patient was a 74-year-old female and diagnosed with stage IV poorly differentiated adenocarcinoma of lung with adrenal gland, hilar, and mediastinal lymph nodes metastasis (T2N2M1) in May 2016. As ARMS-PCR showed EGFR 19 deletion, she got gefitinib treatment since July 2016, with a PFS of 18 months. In December 2017, pleural metastasis, bilateral supraclavicular lymph node metastasis occurred with progression of mediastinal and bilateral hilar lymph node metastasis as well. So, she switched to carboplatin, pemetrexed and bevacizumab treatment for four cycles and followed by bevacizumab monotherapy for 13 cycles until July 2019 when cerebral infarction occurred. On September 2019, NGS testing of ctDNA showed a MET kinase domain mutation (p.H1094Y) along with an EGFR exon 19 deletion (p.E746_A750del). So, the patient started crizotinib as the third-line treatment on October 2019. Though she reached a PR in the first month, the lung lesion and hilar lymph node metastasis progressed on January 2020. Supraclavicular lymph node biopsy was further tested with NGS, a secondary MET p.D1228H, which indicated the resistance of crizotinib was identified (Figure 4(c)). Later, the patient was treated with carboplatin, pemetrexed with a best response of PR, but finally the disease progressed and the patient passed away in January 2021.
We also identified a novel exon 14 skipping mutation in a patient who were treated with immune checkpoint inhibitor therapy. This patient was a 77-year-old male, and he was diagnosed with stage IV lung adenocarcinoma with left hilar, mediastinal, tracheal node, and right humerus metastasis (T4N3M1b). Biopsy of the right supraclavicular lymph nodes showed poorly differentiated lung adenocarcinoma. As immunohistochemistry (IHC) showed ALK (-), ROS1 (-), RT-PCR of EGFR showed no actionable mutation (EGFR wild type), the patient was enrolled in a phase III clinical trial (CTR20170064). The patient was assigned to the group of combined atezolizumab (PD-L1 inhibitor) and chemotherapy group and started atezolizumab 1200 mg, pemetrexed, and cisplatin on March 2018. On April 2018, computed tomography (CT) scan showed the shrunk of the left hilar, mediastinal, tracheal node, and the primary lesion. PR was reached and maintained until January 2019. In February 2019, the primary lung lesion became enlarged (PD) after he switched to atezolizumab 1200 mg monotherapy for 1 month. Therefore, docetaxel was chosen as the second-line therapy. However, inguinal lymph nodes were enlarged and MRI showed new brain metastasis (PD) 1 month later. To explore other potential therapies, a comprehensive genomic profiling of the inguinal lymph nodes (Geneplus-Beijing, Beijing, China) was performed (Figure 5(a) and (b)). A novel MET ex14 alteration (c.3028+3_3025+5delATAinsTTT) was identified in the inguinal lymph nodes (Figure 5(b)). As the MET gene copy number was 2.8, an additional IHC of MET showed high expression of MET (3+, H-score 280) (Figure 5(c)). RNA sequencing also confirmed the exon 14 skipping of this novel mutation (Figure 5(d)). Then he was enrolled in a phase II clinical trial for a MET inhibitor savolitinib (CTR20160581). He had a tumor reduction of 56.5% in the lesion of left lung, and a significant shrunk of the brain lesion and bilateral inguinal lymph nodes as well (PR) 6 weeks after savolitinib treatment (Figure 5(e) and (f)). At date cut-off, September 2021, the patient remains on savolitinib treatment.

Genetic and clinical information from the patient (M098) before and after MET inhibitor treatment. (a) Hematoxylin and eosin-stained biopsy. (b) Mutation of the MET mutation: MET (NM_000245.2: c.3028 + 3_3025+5delATAinsTTT). (c) Immunohistochemistry (IHC) staining image of the biopsy of the recurrent left lung sample. MET IHC (3+), 90, H-Score:280. (d) Schematic representation of the MET mutation confirmed by RNA sequencing. (e) Chest and (f) brain computed tomography (CT) revealed the clinical response to MET inhibitor, savolitinib. Upper panel: before savolitinib treatment; lower panel: 1 month after savolitinib treatment.
Discussion
MET is a receptor tyrosine kinase that drives oncogenesis in many different types of tumors. Most studies were conducted in advanced stages for the clinicopathological characteristics and predictive implications of MET exon 14 skipping mutations and MET amplification before treatment of NSCLC.15,40 Data concerning the overall MET alterations in both treatment-naïve and treated patients is scanty. To our knowledge, the current study represents the largest cohort for the comprehensive assessment of MET nonactionable mutations, MET CNG, MET exon 14 skipping mutations, MET kinase domain activating mutations, and MET rearrangement in both early and advanced, treatment-naïve, and treated NSCLC patients.
Among the 642 MET mutations identified in the 564 NSCLC patients, 330 (50.9%) were actionable mutations. MET CNG, the most common actionable mutation was not an early event of tumorigenesis, but enriched in advanced stage and especially those treated patients as an acquired resistant mechanism of EGFR-TKI. This was consistent with previous report that EGFR-TKI therapy may induce the structural variant of MET,5,25 especially for CNG. The lower occurrence of MET CNG may also contribute to the lower percentage of actionable mutation in stage I–IIIa than that of stage IIIb–IV patients. The 117 MET exon 14 skipping mutations had a similar distribution pattern with previously reported data.9,41 We also observed that MET exon 14 skipping occurred with MET CNG in 13 patients, 32 and the allele frequency of MET exon 14 skipping was significantly higher in the patients with MET CNG (Figure 2(d)). Thus, we speculated that most CNG of MET may occur in the allele with MET exon 14 skipping. Although activating in one single allele of an oncogene is believed to be sufficient to drive tumorigenesis, concurrent mutation and CNG have been reported in EGFR, KRAS, and other oncogenes. Such mutant allele specific imbalance of oncogenes has been noted in human cancers, 42 though the underlying mechanisms remain to be elucidated.
In our cohort, patients pretreated with EGFR/ALK/ROS1 TKI (n = 111) had different clinical characteristics from other stage IIIb–IV patients (n = 346) including age, gender, and smoking statue (Table 1). The MET alteration profile was significantly different as well, presenting as higher frequency of MET CNG, MET GOF mutation, but lower frequency of MET exon 14 skipping as expected. 3 These MET alterations contributed to the resistance of EGFR/ALK/ROS1 TKI as previous reports.24,43,44 EGFR-TKI-treated patients had significantly higher percentage of MET CNG, and higher copy numbers of MET compared than treatment-naïve advanced patients. Interestingly, 8 of the 10 patients with GOF mutation of MET in the kinase domain were previously treated with EGFR-TKI, with two of them were concurred with EGFR T790M mutation. However, for the 3 MET exon 14 skipping identified in EGFR-TKI-treated patients, two were concurred with EGFR T790M mutation. This indicated that high-level MET CNG or MET GOF mutation was more common than MET exon 14 skipping in EGFR-TKI-resistant setting.
For the 22 patients treated with crizotinib or savolitinib, an overall objective response rate of 68.1% and mPFS of 10 months were reached. This mixed efficacy data were similar to those reported in MET-amplified patients and in MET exon 14 skipping patients treated with crizotinib.12,45 The three patients with combined EGFR-TKI and MET-TKI were all in the crizotinib group, and the PFS of these three patients were 3, 3, and 8 months respectively. Maybe it contributed to the general shorter mPFS in the crizotinib group than the savolitinib group. However, due to the heterogeneity of previous treatment and MET alterations, this information should be interpreted with caution, even though studies have showed the great efficacy of savolitinib in combination with the third-generation EGFR-TKI osimertinib in patients progressed to prior EGFR-TKI treatments.21,22
Our study has some limitation. As a retrospective study of the MET mutation spectrum, we could not get access to most prognosis data. However, our study indeed showed that MET actionable mutation could occur in heavily treated patients and still responded well to MET-targeted therapy, and double MET GOF mutation may occur as a required mechanism to EGFR-TKI. Second, there was no IHC data to study the correlation between MET IHC and DNA alterations. However, it has been found that for the detection of high-level MET amplification and polysomy, IHC was highly sensitive and had a 100% negative predictive value. 32
In conclusion, this is the first study parallel comparing MET status across different stages and different treatment history of NSCLC. It demonstrated that MET actionable mutations including CNG, exon 14 skipping and GOF mutations occurred differently in different clinical scenario. MET inhibition may be still effective in heavily treated patients with actionable mutations.
Supplemental Material
sj-docx-2-tam-10.1177_17588359221112474 – Supplemental material for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting
Supplemental material, sj-docx-2-tam-10.1177_17588359221112474 for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting by Ai Xinghao, Yongfeng Yu, Jun Zhao, Wang Sheng, Jing Bai, Zaiwen Fan, Xuemei Liu, Wenxiang Ji, Rongrong Chen and Shun Lu in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-png-1-tam-10.1177_17588359221112474 – Supplemental material for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting
Supplemental material, sj-png-1-tam-10.1177_17588359221112474 for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting by Ai Xinghao, Yongfeng Yu, Jun Zhao, Wang Sheng, Jing Bai, Zaiwen Fan, Xuemei Liu, Wenxiang Ji, Rongrong Chen and Shun Lu in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-xlsx-3-tam-10.1177_17588359221112474 – Supplemental material for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting
Supplemental material, sj-xlsx-3-tam-10.1177_17588359221112474 for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting by Ai Xinghao, Yongfeng Yu, Jun Zhao, Wang Sheng, Jing Bai, Zaiwen Fan, Xuemei Liu, Wenxiang Ji, Rongrong Chen and Shun Lu in Therapeutic Advances in Medical Oncology
Supplemental Material
sj-xlsx-4-tam-10.1177_17588359221112474 – Supplemental material for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting
Supplemental material, sj-xlsx-4-tam-10.1177_17588359221112474 for Comprehensive analysis of MET mutations in NSCLC patients in a real-world setting by Ai Xinghao, Yongfeng Yu, Jun Zhao, Wang Sheng, Jing Bai, Zaiwen Fan, Xuemei Liu, Wenxiang Ji, Rongrong Chen and Shun Lu in Therapeutic Advances in Medical Oncology
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.