
Abstract
Liposarcoma (LPS) is a common soft tissue sarcoma that encompasses diverse subtypes of well-differentiated/dedifferentiated, myxoid/round cell, and pleomorphic LPS. There is heterogeneity among the various LPS types with regard to prognosis, molecular pathogenesis, and response to treatment. Well-differentiated (WDLPS) and dedifferentiated liposarcoma (DDLPS) are most common types, which share common genetic alteration of chromosome 12q13-15 amplification resulting in amplification of oncogenes, including MDM2 (Mouse double minute 2), CDK4 (cyclin-dependent kinase 4), and HMGA2 (High mobility group protein AT-hook 2). Despite sharing the same molecular alteration, DDLPS has a worse prognosis, with a higher recurrence rate and higher propensity for metastases compared to WDLPS. Here we provide an overview of the LPS treatment landscape focusing on recent developments in the treatment of DDLPS with a focus on selinexor. Selinexor, a selective inhibitor of XPO1, was recently evaluated in a phase 3 trial, the first prospective randomized trial in DDLPS, and we discuss its efficacy in context of other available agents for DDLPS.
Keywords
Introduction
Soft tissue sarcoma (STS) encompasses a heterogeneous group of tumors arising from mesenchymal structures. Liposarcoma (LPS) is one type of this rare group of tumors that originate from adipocytes and is reported to be about 15%–20% of all STS and the age-adjusted incidence rates are 1.08 and 1.01 per 100,000 person-years in 2001 to 2016 from the SEER and CNPCR database.1,2 Three main biologic subtypes of LPS include well-differentiated/dedifferentiated, myxoid/round cell, and pleomorphic. There is heterogeneity within the group of LPS with regards to prognosis, molecular pathogenesis, and response to treatment.
Well-differentiated (WDLPS) is considered low grade and has the best prognosis, pleomorphic liposarcoma (PLS) the worst, and myxoid/round-cell liposarcoma (MRCLS) has an intermediate prognosis. MRCLS accounts for about 30%–40% of LPS and around 53% of cases have a round cell component. 3 Presence of more than 5% round cell transformation denotes poorer prognosis with a higher rate of metastasis and a drop in the overall survival at 5 years from 90% to just 50%. 2 MRCLS is characterized by a chromosomal translocation and in 95% of cases this results in the FUS-CHOP gene fusion that serves as a transcription activator. PLS is a high-grade tumor with a reported recurrence rate and systemic relapse rate of 59% and 25%, respectively. 4 This rare type of LPS that accounts for only 10% of all LPS, has no pathognomonic molecular aberration, but frequently has p53 mutations.4,5
WDLPS and dedifferentiated liposarcoma (DDLPS) are most common types, which share common genetic alteration in chromosome 12q13-15 amplification resulting in amplification of oncogenes including MDM2 (Mouse double minute 2), CDK4 (cyclin-dependent kinase 4), and HMGA2 (High mobility group protein AT-hook 2).6,7 Despite sharing the same molecular alteration, DDLPS has a higher recurrence rate, metastatic potential, and higher overall mortality rate documented as high as 50%–70%. WDLPS mostly occurs in extremities (75%), where they are often called atypical lipomatous tumor (ALT), and around 25% arise in the retroperitoneum, on the other hand, majority of DDLPS (around 75%) arise in the retroperitoneum. 5 DDLPS will often have a well-differentiated component, and although DDLPS is more commonly found de novo, 10%–20% of WDLPS eventually recur as or progress to DDLPS.8–10
Treatment of LPS depends on location, stage, and size, which together determine resectability, and also the subtype, which determines sensitivity to systemic therapy. Surgery remains the main treatment for localized LPS, while systemic treatment is used selectively in the adjuvant/neoadjuvant setting for high grade LPS that are over 5 cm. Systemic therapy is the mainstay for metastatic disease. Current standard systemic therapies include doxorubicin-based regimens as front-line therapy, with gemcitabine-docetaxel, trabectedin, and eribulin used in later lines. The different LPS have variable sensitivity to these chemotherapies; MRCLS being the most sensitive to doxorubicin-based regimens and trabectedin, while pleomorphic liposarcoma has a longer overall survival (OS) benefit with eribulin.11–13 WDLPS/DDLPS remain a challenge given their higher prevalence and lower response rate with standard chemotherapy options. Purely well-differentiated tumors have no documented benefit from the current systemic options and their remains a paucity of active agents for DDLS. The availability of novel agents targeting the MDM2-p53 oncogenic axis have led to newer agents been investigated in this space. Selinexor, a selective inhibitor of XPO1 was recently evaluated in a phase 3 trial: the first prospective randomized trial in DDLPS. Here we will review available data in context of other agents in DDLPS and determine how this agent with a marginal, but statistically significant improvement in PFS, might fit in.
Currently available systemic therapies
Table 1 summarizes systemic therapies that have been used in WDLPS/DDLPS.
A summary of treatment choices and outcomes in unresectable or metastatic dedifferentiated liposarcoma.

DDLPS, dedifferentiated liposarcoma; DTIC, dacarbazine; mOS, median OS from chemotherapy initiation; mPFS, median PFS; ORR, overall response rate; WDLPS, Well-differentiated liposarcoma.
Statistically significant different as compared to control group of the study.
Median overall survival for patients who did not cross over to selinexor.
Estimate from the results in weeks; PFS per report were 17.9 weeks and 30.4 weeks for palbociclib and abemaciclib, respectively.
Traditional chemotherapy
As for most STSs, first-line systemic therapy for DDLPS is doxorubicin-based chemotherapy. A large phase 3 randomized controlled trial: EORTC 62012 compared single agent doxorubicin versus combination of doxorubicin and ifosfamide in first line locally advanced, unresectable or metastatic STS. 24 This combination regimen demonstrated improved overall response rate (ORR) and PFS but not overall survival (OS). The median OS was 12.8 and 14.3 months for single doxorubicin and the combination (HR 0.83, 95% CI: 0.67–1.03), respectively. The combination regimen had more side effects and lead to dose reduction in 32% and discontinuation in 18%. 24 In the study, LPS comprised 11% of STS in the doxorubicin arm and 14% in the combination arm, though further subtype specific efficacy was not reported. Efficacy data regarding DDLPS specifically has been evaluated in retrospective studies. A multi-institutional study from Europe with 208 WDLPS/DDLPS patients, reported first line systemic therapy response rate of 12%, median PFS of 4.6 months, and median OS of 15.2 months. 14 Most of the cases (82%) had DDLPS, 82% (171 of 208 patients) received anthracycline-containing regimens, and 41% received combination therapy. This study reported a higher ORR with an anthracycline-containing regimen compared to non-anthracycline (15% vs 0%) and combination chemotherapy compared to single (18% vs 7.5%). Data from the study also reported no difference in ORR between WDLPS and DDLPS (13% and 12%) though WDLPS numbers were small, and it was not clear if the WDLPS diagnosis was at baseline or before the start of chemotherapy. Other studies have reported lack of response in WDLPS with chemotherapy, compared to DDLPS. 25 A report from a single institute experience containing 82 evaluable patients, revealed an ORR to front-line therapy of 21% in DDLPS, with a higher ORR with doxorubicin combinations (30.2% with doxorubicin/ifosfamide and 25% with doxorubicin/dacarbazine) compared to single agent doxorubicin (0%). 15 This study excluded patients with exclusively WDLPS histology. Patients in the recurrent/metastatic setting had a median PFS of 4 months (95% CI: 3–7 months) and median OS from initiation of chemotherapy of 25 months (95% CI: 18–31 months). 15
Traditionally, gemcitabine combined with docetaxel is used in the second line, based again on studies in STS. SARC 002 was a phase 2 trial comparing outcomes between single agent gemcitabine and gemcitabine-docetaxel in metastatic STS. 16 Combination gemcitabine-docetaxel had a superior ORR (16% vs 8%), median PFS (6.2 months vs 3 months), and median OS (17.9 months vs 11.5 months), compared to gemcitabine. LPS patients made up 24.5% of the gemcitabine arm and 11% of the gemcitabine-docetaxel arm. Of the WDLPS/DDLPS patients none were noted to have a response in either arm, while 4 out of the 5 evaluable patients had stable disease (SD) in the combination arm.16,26 Another clinical study of gemcitabine-docetaxel was GeDDis trial comparing it with single-agent doxorubicin in the front-line treatment of STS. 27 The results revealed no significant difference in PFS and OS, with the toxicity profile favoring single agent doxorubicin. This study included 8 (6.2%) WDLPS/DDLPS patients in the doxorubicin arm and 5 (3.9%) in the gemcitabine-docetaxel arm.26,27
In later lines of treatment in DDLPS, trabectedin or eribulin are often used as they are FDA approved for the treatment of LPS based on survival benefit over dacarbazine. A phase 3 randomized multicenter clinical trial of trabectedin compared to dacarbazine in metastatic LPS or leiomyosarcoma showed a superior PFS (HR 0.55, p < 0.001) but no difference in OS. 19 This trial comprised of 27% (140/518 patients) LPS patients in both arms that included MRCLS, PLS as well as DDLPS. In a preplanned subgroup analysis for LPS, median PFS in trabectedin arm was 3.0 months, significantly improved from 1.5 months in dacarbazine arm (HR 0.55, 95% CI: 0.34–0.87, p = 0.009). In DDLPS subgroup, median PFS was 2.2 months in trabectedin (vs 1.9 months in dacarbazine arms, HR: 0.68, 95% CI: 0.37–1.25) while median PFS for MRCLS and PLS was 5.6 months (vs 1.5 months in dacarbazine) and 1.5 months (vs 1.4 months in dacarbazine), respectively.
In a similar phase 3 trial, eribulin demonstrated modest activity in L-type sarcomas (LPS and leiomyosarcoma) with significantly increased OS but not PFS compared to dacarbazine. 18 Of the 452 patients, 143 had LPS and subgroup analysis in LPS demonstrated a statistically significant benefit in PFS and OS with eribulin. OS in this subgroup increased from 8.4 months to 15.6 months (HR: 0.51; 95% CI: 0.35–0.75; p = 0.001) and PFS increased from 1.7 months to 2.9 months (HR 0.52; 95% CI: 0.35–0.78; p = 0.0015) in the dacarbazine vs eribulin arms, respectively. While adverse events were similar between arms. Only one patient with PLS had a partial response (PR) to eribulin and the PFS was 4.4 months (vs 1.4 months in dacarbazine) for PLS, 2.0 months (vs 2.1 months in dacarbazine) in DDLPS, and 2.8 months (vs 1.4 months in dacarbazine) in MRCLS. 11
Targeted therapy
There are currently no approved targeted therapies in LPS. Off label use of pazopanib and CDK4 inhibitors like palbociclib and abemaciclib are occasionally used, supported based on available published studies.
Pazopanib is an oral multitargeted tyrosine kinase inhibitor (TKI) against VEGFR (Vascular Endothelial Growth Factor Receptor) and PDGFR (Platelet Derived Growth Factor Receptor) that is currently FDA approved for non-adipocytic STS. The approval was based on a significant PFS advantage compared to placebo in a multi-center phase 3 ‘PALETTE’ (PAzopanib expLorEd in sofT TissuE sarcoma) study in 2012. 28 LPS was excluded from this study due to failure to meet the prespecified efficacy cut-off in the preceding phase 2 EORTC (European Organization for Research and Treatment of Cancer) study 62043. However, central histopathologic review later reclassified 2 patients increasing the progression-free rate (PFR) at 12 weeks from 17.6% to 26% (5 of 19 patients) in LPS. This number would have met the criteria to continue recruiting patients in the cohort. Thereafter, a single arm phase 2 study was conducted to further define the clinical benefit of pazopanib in LPS with PFR as the primary endpoint. 22 The PFR at 12 weeks was 68.3% (95% CI: 51.9%–81.9%) with an ORR of 2.4% and clinical benefit rate (CBR) of 44%, which met the prespecified cut off for activity. Median PFS was 4.4 months and median OS was 12.6 months (95% CI: 8.5%–16.2 months). LPS subtype data is not available. Based on the landmark study by the EORTC evaluating appropriate endpoints for STS for noncytotoxic drugs in clinical trials, a 3-month PFR of ⩾ 40% suggests an active drug in second-line. 29
MDM2 and CDK4 are overexpressed in almost all WDLPS and DDLPS. MDM2 is a negative control of p53 while CDK4 is involved in Rb phosphorylation and leads to G1-2 phase progression and cell proliferation. A phase 2 prospective study of Palbociclib, a CDK4/6 inhibitor, in WDLPS/DDLPS led to complete response in 1 patient out of a total of 57 evaluable patients, with a 12-week PFS of 57%, and a median PFS of 17.9 weeks.23,30 Abemaciclib, which is more selective for CDK4, has shown more promising results in a phase 2 study. 23 Abemaciclib 200 mg twice daily demonstrated 1 PR out of a total of 30 patients, 12-week PFS of 76%, and median PFS of 30.4 weeks. A larger randomized study with abemaciclib in DDLPS is underway (NCT04967521).
Immunotherapy
Role of immunotherapy in liposarcoma is still under investigation. A study in DDLPS cell lines had demonstrated PDL1 ⩾ 1% protein expression in 21.9%. 31 The initial study of Pembrolizumab in the SARC028 study reported 2 out of 10 LPS patients had response to the therapy, but after enrolling additional patients, the response was lower (10%). 32 The role of checkpoint inhibitor combinations are currently being explored in LPS (NCT03899805, NCT03307616, NCT04668300).
Selinexor
Selinexor is the first generation of selective inhibitors of nuclear export (SINEs) and currently the first to receive approval from the FDA for cancer therapy. Selinexor reversibly and specifically inhibits XPO1, a nuclear exportin. Here we will review in detail the mechanism of action and available clinical data.
Selinexor mechanism of action
In human cells, transportation of molecules in and out of nucleus happens via the nuclear pore complex (NPC). Molecules larger than 40 kDa (kilodalton) including RNAs need energy-dependent active transport mediated mainly by the karyopherin family of proteins. 33 This family of protein consists of importins and exportins. XPO1, also known as chromosomal region maintenance 1 (CRM1) is the only exportin which can recognize a leucine-rich hydrophobic nuclear export signal (NES). After it recognizes its cargo proteins, XPO1 exports them through NPC by energy from a Ran energy gradient system (Figure 1). NES-containing proteins have been discovered for at least 220 proteins. Among these, several tumor suppressor proteins (TSPs) are included, for example, p53, p73, BRCA1/2, IκB, p21, p27, and FOXO transcription factors.33–35 In addition to tumor suppressor proteins, XPO1 is also required for the export of cap-binding protein eIF4e (eukaryotic translation initiation factor 4e). 35 This protein exports mRNAs of many oncoproteins from the nucleus for translation. These oncoproteins include c-MYC, MDM2, and Cyclin D. Since the function of TSPs require their presence in the nucleus for cell cycle regulation and DNA damage recognition, overexpression and function of XPO1 can inactivate TSPs. Also, XPO1 overexpression can lead to promotion of oncoprotein syntheses.

Physiologic function of XPO1.
XPO1 inhibitors have been developed since the early 1980s. The first drug was leptomycin B, an antifungal agent extract from a strain of Streptomyces.36,37 However, further study was halted due to the off-target side-effects from the drug and a narrow therapeutic window. Currently XPO1 inhibitors being studied involve PKF050-638, CBS9106 and SINEs. Among SINEs, selinexor is the first that has an FDA-approved use in clinic. Selinexor covalently binds to Cys528 in the NES-binding pocket on the convex outer surface with a slowly reversible effect allowing normal cell to survive and causing less side effects than leptomycin B. Binding to the NES-binding pocket prohibits cargo proteins to bind and subsequently causes TSPs and oncoprotein mRNAs to be retained in the nucleus (Figure 2). Theoretically these effects are expected to cause apoptosis selective to a damaged genome, as in malignant cells.

Mechanism of action of selinexor/SINE.
Selinexor clinical data
Alteration of XPO1 functions were originally observed with higher expression in several hematologic malignancies and almost all solid tumors, and recurrent XPO1 gene mutations were noted in primary mediastinal B-cell lymphoma (PMBL), Hodgkin’s lymphoma, and CLL. 38
In a phase 1 study, selinexor demonstrated a response rate of 4.5% (7/157) in advanced solid malignancies unresponsive to available therapies or for which no standard therapy existed. 35 Of the patients included, 31% had colorectal cancer, 11% had HN-SCC, 11% had prostate cancer, 8% had melanoma, 6% had pancreatic cancer, and 5% had sarcoma.
In phase 1 studies of acute myeloid leukemia (AML), treatment with single agent selinexor had a response rate of 14% (out of 81) in heavily pretreated patients, while combination therapy with high dose cytarabine and mitoxantrone in newly diagnosed or relapsed/refractory AML had achieved overall response rate as high as 70%.39,40 Although patients achieved response with an encouraging PFS and OS in phase 1, a phase 2 study, SOPRA (Selinexor in Older Patients with Relapsed/Refractory AML), failed to demonstrate significant improvement in OS compared to physician’s choice treatment. 41 Selinexor in combination with other drugs showed higher ORRs of 40%–-70% in many phase-1 studies, the highest seen in combination with high-dose cytarabine and mitoxantrone.42–44 Further studies are ongoing in AML.
Selinexor has been approved by the FDA for treatment of multiple myeloma (MM) in 2 settings based on the results of the BOSTON trial and the STORM trial (Table 2). In the phase 3 BOSTON trial, the triplet combination (Selinexor, bortezomib, dexamethasone) revealed a statistically improved progression free survival (PFS) from 9.46 months to 13.93 months (95% CI: 0.53–0.93, p = 0.0075) compared to standard therapy with bortezomib and dexamethasone. 45 The STORM (Selinexor Treatment of Refractory Myeloma) trial, a phase 2b study, also showed benefit of selinexor in triple-class refractory myeloma where no available treatments have proven benefit, defined as disease refractory to proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies. 46 Of a total of 122 patients, 32 patients (26%) had response to the selinexor with a median duration of response of 4.4 months (95% CI: 3.7–10.8). As a result of these trials, selinexor has been approved for use in these two settings (Table 2).
Current FDA approved indications for selinexor.

Diffuse large B-cell lymphoma (DLBCL) has been shown to have overexpression of XPO1 and this is related to a poor prognosis. Also, in most B-cell lymphomas eIF4e is overexpressed as well. 48 This protein carries mRNAs of oncoproteins for nuclear export via XPO1 to the cytoplasm for translation. In preclinical models SINE demonstrated XPO1 accumulation in the nucleus and apoptosis of lymphoma cells independent of p53.47,48 Single agent selinexor had a response rate of 28% with 12% complete response in DLBCL patients in the phase 2b SADAL trial (Table 2). 47 Median OS for patients with response was not reached (29.7-NE) while median OS was 4.3 months (3.0–5.4) in patients who had progressive disease or were not evaluable for response. This data led to the approval in refractory DLBCL.
Selinexor as a treatment of liposarcoma
Preclinical data
In vitro and in vivo preclinical models of sarcoma indicated the effects of selinexor in decreasing cell proliferation and inducing cell cycle arrest and apoptosis. A study confirmed that selinexor inhibited XPO1 at protein level and the reduction in XPO1 protein in LPS cells led to inhibition of cell proliferation via cell cycle arrest (increase in G1 phase while decrease in S and G2M phase) and apoptosis. 36 Studies in LPS cell lines with MDM2 amplification showed selinexor increased p53 and p21 expression at protein level without effect to p53, MDM2, and CDKN1A mRNA levels and induced G1 arrest and tumor cell apoptosis independent of p53 and RB status. 49 In the latest study in DDLPS PDX models, selinexor showed a moderate antitumor activity in induction of apoptosis consistently higher than doxorubicin. The treatment response of both drugs was not influenced by the level of MDM2 and CDK4 gene amplification. 50
Clinical data
Although the exact mechanism of action of selinexor toward LPS was yet to be clarified, early phase clinical studies suggested selinexor activity in DDLPS. In a phase 1B clinical study of selinexor in sarcoma with a total of 52 patients, 33% experienced SD for 4 months or longer. 51 However, more patients in DDLPS demonstrated reduction in target lesion than other subtypes of sarcoma. Of the 15 DDLPS patients, 40% had reduction in target lesions and 47% had SD for 4 months or longer. This study also evaluated growth modulation index (GMI) which is the ratio of time to tumor progression (TTP) on selinexor versus TTP with immediate prior therapy. According to previous studies, a GMI ⩾ 1.33 correlates with drug activity and associated with OS improvement in advanced STS.51–53 Of note, in this phase 1B study, 54% from total of 13 evaluable DDLPS patients had GMI ⩾ 1.33 whereas 39% of the 41 evaluable total STS patients had GMI ⩾ 1.33. 51
These promising data in DDLPS led to the phase 2/3 randomized double blind, placebo controlled cross-over study (SEAL). 54 The SEAL study evaluated efficacy of single agent selinexor in advanced unresectable DDLPS with progressive disease after 1 or more prior systemic therapies. The phase 2 part used PFS by WHO criteria as the primary end point and by RECIST version 1.1 as pre-specified analyses. The results of the first 56 patients were presented in 2018, where the median WHO-PFS of selinexor and placebo revealed no difference (1.4 month in both arms, HR: 0.92; 95% CI: 0.52–1.63) while the median PFS by RECIST version 1.1 was 5.6 and 1.8 months (HR = 0.64; 95% CI: 0.31–1.32, p = 0.21) respectively. The improvement trend in PFS by RECIST criteria supported continuation to phase 3. A comparison analyses between WHO and RECIST 1.1 criteria revealed WHO response criteria lead to premature determination of asymptomatic progression events involving small lesions while overall tumor burden was stable.
In the SEAL phase 3 study, 285 patients who received 2-5 lines of prior therapy with radiologic progression noted within the prior 6 months were randomized to selinexor or placebo in a 2:1 allocation. 21 The primary endpoint was PFS and secondary endpoints were TTP, ORR, duration of treatment (DOT), and time to next treatment (TTNT). Progression of disease was based on independent radiology review using RECIST 1.1 criteria based on the recommendation from phase 2. Results were presented at the Connective Tissue Oncology Society (CTOS) 2020 annual meeting based on 209 PFS events. The independent radiology review determined PFS in the selinexor arm (2.83 months) was improved compared to placebo (2.07 months), and this result was statistically significant with a hazard ratio of 0.70 (95% CI: 0.52–0.95) and a two-sided p-value of 0.0228. The 6 and 12 months PFS rates were 23.9% and 8.4% in selinexor versus 13.9% and 2% in the placebo arm. The median OS was not statistically significantly different by intention to treat analysis (ITT), being 9.99 and 12.91 months (HR 1.0039, 2-sided p-value 0.9836) in selinexor and placebo arms, respectively, with crossover rate from placebo to selinexor documented at 58%. The ORR was 2.7% for selinexor and 0% for placebo. Further analysis showed patients with ⩾ 15% disease burden reduction was 7.5% noted in selinexor and 0% in placebo arm.
Although the main clinical outcomes publication of the SEAL trial including the correlatives are awaited, the secondary end point of HRQoL outcomes measured by EORTC QLQ-C30 have been published recently. 55 Quality-of-life assessments including physical functioning, role functioning, pain, and global health/QoL were performed as per protocol. The patients in selinexor group had reported lower pain rated and slower worsening of pain compared to placebo group with the mean difference in pain score were -12.18 and -14.24 at day 127 and 169, respectively (p = 0.032 and p = 0.031), while other domains did not significantly differ between the 2 groups.
Dosing and side effects
In the phase 1B study of selinexor in advanced solid tumors, the safety, maximum-tolerated dose (MTD) and recommended phase 2 dose (RP2D) of selinexor were evaluated. 36 The dose-limiting toxicities (DLT) included grade 3 fatigue, nausea and vomiting, hyponatremia, acute cerebellar syndrome, and anorexia. The study established the RP2D of selinexor as 35 mg/m2 twice a week or 60 mg fixed dose. At this dose the efficacy was comparable to higher doses with better tolerability.
In SEAL phase 2/3 clinical study, selinexor 60 mg fixed dose regimen was given twice weekly in 42 day-cycles until progression of disease or intolerability. 54 Most patients got early intervention and prophylaxis for nausea and anorexia as part of the protocol treatment. The result from the phase 2 part which was unblinded for 51 patients, 24 on selinexor and 37 on placebo, reported common grade 1/2 adverse events (selinexor vs placebo) of nausea (85% vs 31%), anorexia (62% vs 14%), and fatigue (58% vs 31%) and grade 3/4 adverse events of hyponatremia (15% vs 0%), anemia (15% vs 7%), and thrombocytopenia (12% vs 0%). Overall toxicities were reported as manageable with treatment discontinuation due to toxicity 10.2%. 21 The full side effect profile from this study is yet to be published.
We have summarized available treatment-related adverse events of single agent selinexor at the currently recommended dose from available clinical trials and presented them in Table 3. The limited data set of side effects from the DDLPS study matches the prior data from the refractory sarcoma phase 1B trial. Majority of treatment-related adverse events (TRAE) presented in advanced sarcoma were grade 1 and 2 which were nausea, contitutional symptoms, and hyponatremia (Table 3). While the phase 2B study in DLBCL reported more grade 3-4 neutropenia and thrombocytopenia but less fatigue and weight loss. Acute cerebellar syndrome with associated ataxia and dysarthria, one of the dose-limiting toxicities seen in a phase 1 study in advanced solid tumor at 85 mg/m2 dose (grade 4 but reversible over 6 weeks) was not seen at the 60 mg twice a week dose.
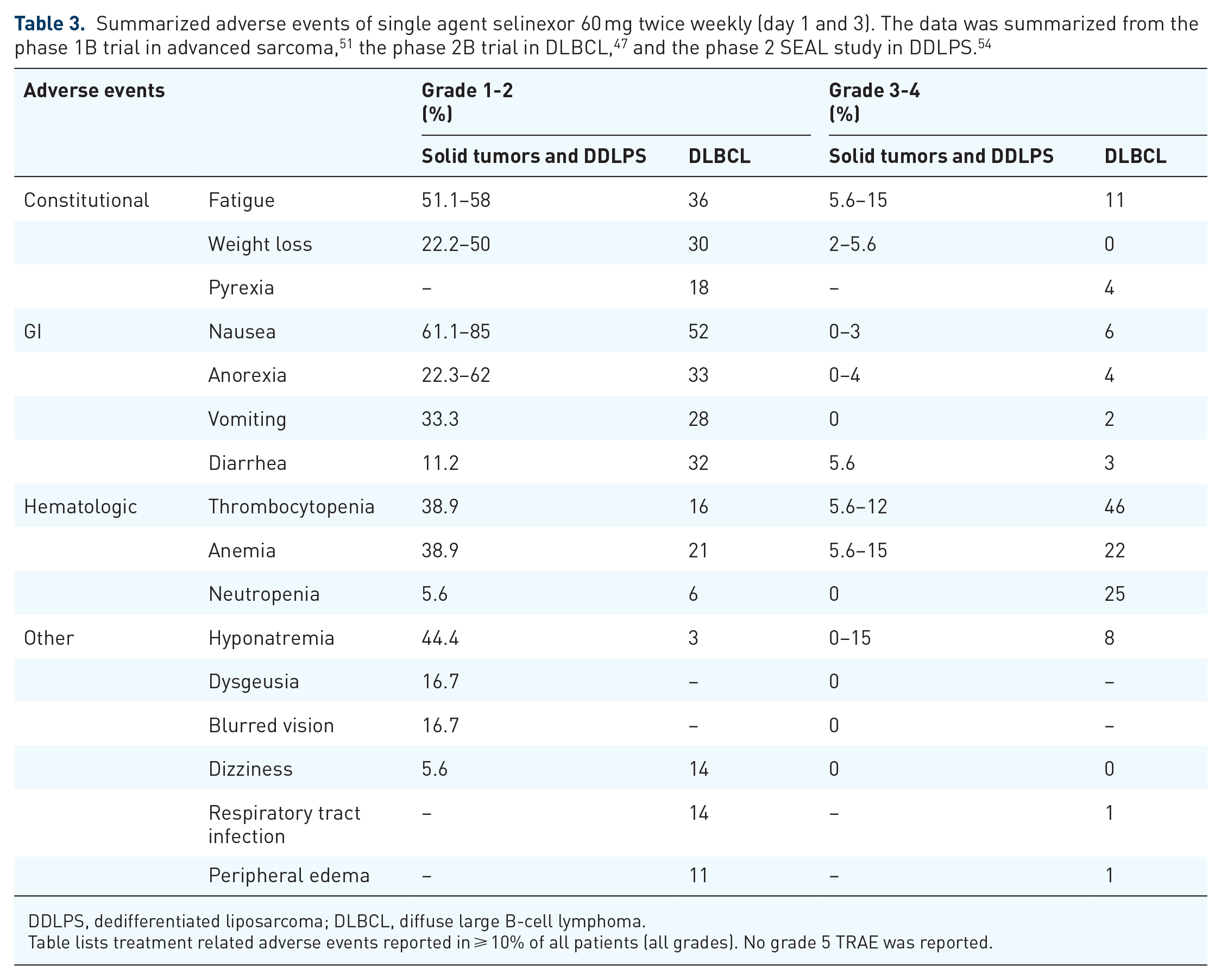
DDLPS, dedifferentiated liposarcoma; DLBCL, diffuse large B-cell lymphoma.
Table lists treatment related adverse events reported in ⩾ 10% of all patients (all grades). No grade 5 TRAE was reported.
In conclusion, TRAE of selinexor 60 mg twice weekly on day 1 and 3 has proved to be manageable, with supportive care and early intervention for nausea and anorexia being key in improving tolerability and allowing patients to remain on study. 36
Selinexor: biomarker data
A variety of biomarkers have been studied in selinexor studies so far, including XPO1 mRNA, p53 mutational status, MDM2, and CDK4 amplification level. Increased XPO1 mRNA in peripheral blood mononuclear cell (PBMC) after administration of selinexor has been shown to associate with appropriate drug engagement and activity toward target protein, XPO1. 39 However, a small phase II study of selinexor in triple-negative breast cancer did not find any correlation of the mRNA induction with patients who derived clinical benefit. 56 Data from phase I dose escalation study of selinexor in solid cancer demonstrated RAS and AKT pathway activation in colon cancer had a higher percentage of DCR of > 3 months compared to the no RAS/PI3 K-AKT mutation group in the 18 patients evaluated. 57 These have yet to be validated in a larger population study.
In DDLPS, only a subset of patients derived benefit from selinexor leading to substantial improvement in PFS and no benefit in OS, highlighting the importance of predictive biomarkers to select the appropriate population who would be most likely to benefit from the treatment. In a recent pre-clinical study using DDLPS patient-derived xenografts (PDXs), the response to selinexor was not dependent on the level of MDM2 and CDK4 amplification. 50 Although the same study found that treatment with the drug induced p53 overexpression and nuclear accumulation, together with survivin down-regulation over time, there is no evidence of these proteins predicting benefit from selinexor in a clinical study so far. The phase IB study in DDLPS had analyzed tumor specimens but did not report any predictive biomarker. 51 Final correlative data from the SEAL study is awaited, however, a preliminary biomarker analysis from SEAL phase 2/3 was presented at the annual American Society of Clinical Oncology (ASCO) 2021 meeting. 58 RNA sequences from biopsies of responsive and resistance lesions revealed lower expression of CALB1 and higher expression of GRM1 in sensitive tumors while no differential expression of the genes was found in placebo-treated patients. These finding will need validation before we can determine their future as patient selection biomarkers.
Summary and future directions
Selinexor is the first oral systemic therapy of LPS to meet its primary end point of improving PFS in a phase 3 clinical study. However, it was a small incremental benefit in improving PFS compared to placebo in patients who had received 2 to 5 prior lines of systemic therapy. No OS benefit was noted, but cross over was allowed. Some patients experienced a reduction in tumor burden, and some derived prolonged benefit. Evaluation for a predictive biomarker for selinexor is ongoing, with expression of CALB1 and GRM1 identified as potential predictive biomarkers in liposarcoma, though more studies are needed for validation. Successfully identifying a predictive biomarker or exploring combination therapies might help increase the magnitude of impact of selinexor in DDLPS.
Given the paucity of effective systemic therapies in DDLPS, successful completion of a phase 3 study in this indication with selinexor adds hope for patients with advanced, unresectable DDLPS. Even though regulatory approval seems unlikely based on the small but significant incremental PFS benefit, it is the largest prospective study in patients with DDLPS and has paved the way for future randomized trials specifically addressing treatments in DDLPS.
Other systemic treatments currently being investigated in LPS include inhibitors of MDM2 and CDK4, given amplifications in these genes are almost universal in WD and DD liposarcoma. A previous phase 1 study of milademetan (DS-3032b, RAIN-32), an MDM2 inhibitor, revealed benefit in DDLPS patients. 59 Among 3 PR from a total of 25 patients diagnosed with LPS or solid tumors with MDM2 amplification, 1 of the patients with PR was diagnosed with DDLPS. The phase 3 study of milademetan has now been initiated with a comparator arm of trabectedin. 60 Another MDM2 inhibitor, BI907828, has also shown promising results in preclinical and an ongoing phase 1 study as monotherapy or in combination with ezabenlimab in advanced solid tumors and liposarcoma (NCT03964233). 61 CDK4 inhibitors (palbociclib and abemaciclib) have shown promising outcomes as previously described.23,30 A study evaluating abemaciclib in comparison to placebo in DDLPS, has recently started accruing (NCT04967521).
Footnotes
Author contributions
Conflict of interest statement
The authors declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article:
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.