
Abstract
Androgen deprivation therapy (ADT) is the foundation of treatment for patients with locally advanced, recurrent and metastatic prostate cancer, most commonly using luteinizing releasing hormone (LHRH) agonists. More recently, a new approach to ADT has emerged with the development of gonadotropin-releasing hormone (GnRH) antagonists, which aim to overcome some of the potential adverse physiologic effects of LHRH agonists. This article focuses on the newest GnRH antagonist, relugolix – a once-daily treatment and the only oral GnRH antagonist that has now been approved for the treatment of advanced prostate cancer. In phase II and III studies, relugolix achieved rapid and sustained castration without the testosterone surge associated with LHRH agonists, thus avoiding the potential clinical consequences of tumor flare and the necessity for concomitant anti-androgen therapy. Relugolix also achieved rapid testosterone recovery, which may potentially reduce ADT-related adverse events and offer opportunities for combination and intermittent therapy strategies. Cardiovascular safety is a particular concern in men with prostate cancer and ADT further increases cardiovascular risk: indeed, LHRH agonists are required to have a drug label warning about an increased risk of cardiovascular disease. Data from the phase III HERO study demonstrate an improved cardiac safety profile for the GnRH antagonist relugolix compared with the LHRH agonist leuprolide, including a significantly reduced risk for a major adverse cardiovascular event. Taken together, the data indicate that relugolix may mitigate some of the cardiovascular concerns surrounding ADT and has the potential to become a new standard of care for men with prostate cancer. In summary, relugolix represents a novel and recently available prostate cancer management strategy, incorporating the mechanistic advantages of GnRH antagonists and the potential benefits of oral administration.
Keywords
Introduction
Since the landmark recognition by Huggins and Hodges that prostate cancer cells are dependent on testosterone, 1 androgen deprivation therapy (ADT) has been foundational for the treatment of patients with locally advanced, recurrent and metastatic prostate cancer.2,3 Leuprolide was the first commercial luteinizing releasing hormone (LHRH) agonist evaluated in advanced prostate cancer and while numerous agonistic analogs of LHRH have been synthesized, only leuprolide, triptorelin, buserelin, nafarelin and goserelin are routinely used in clinical practice. 4 To date, LHRH agonists, alone or in combination with radiotherapy or other agents, remain the mainstay of treatment for advanced, relapsed or metastatic castrate-sensitive prostate cancer. 5
Initial, and to a lesser extent repeat, administration of LHRH agonists results in an acute rise in luteinizing hormone (LH), follicle-stimulating hormone (FSH) and testosterone (Figure 1).6–9 The clinical implications of this acute testosterone surge are still debated, but it may lead to an exacerbation of clinical symptoms (‘tumor flare’) in advanced disease, including increased bone pain, spinal cord compression, ureteral obstruction, urinary retention and even death.9,10 Long-term exposure to an LHRH agonist, however, eventually desensitizes the pituitary receptor and suppresses LH and FSH production and, consequently, testicular production of testosterone,7–9 although FSH is never completely suppressed. 11

Mechanism of action of GnRH antagonists and LHRH agonists.
Antagonists of the gonadotropin-releasing hormone (GnRH) receptor, which have been developed with a view toward overcoming the potential adverse physiologic effects of LHRH agonists, exert a direct and immediate blockade of GnRH/LHRH pituitary receptors, thereby achieving rapid testosterone suppression without a testosterone surge and potential tumor flare (Figure 1).3,6–8 Abarelix, the first commercial GnRH antagonist, showed good efficacy and a safety profile comparable to that of the LHRH agonist leuprolide, but it was impacted by immediate-onset, histamine-mediated systemic allergic reactions in some patients (1.1%). 12 Abarelix has since been withdrawn from the US market for commercial reasons. 13
Degarelix is a third-generation GnRH antagonist commercially available in an injectable formulation for subcutaneous (SC) administration. 14 In a phase III, randomized trial, degarelix showed non-inferiority to a monthly depot of intramuscular (IM) leuprolide. 15 In keeping with its mechanism of action, degarelix achieved faster testosterone suppression than leuprolide and avoided a testosterone surge. Degarelix also suppressed LH and FSH more rapidly than leuprolide and, in contrast to leuprolide, maintained FSH suppression throughout the treatment period. Injection-site reactions were the most frequent adverse events (AEs) with degarelix and were much more frequent with degarelix than leuprolide (40% versus <1%; p < 0.001). Other frequent AEs with degarelix and leuprolide were generally associated with testosterone suppression.
AEs associated with testosterone suppression are unavoidable with ADT and include not only acute events, such as hot flashes, loss of libido and erectile dysfunction, but also more chronic and less well-characterized events, including fatigue, changes in body composition (gynecomastia, weight gain, reduced muscle mass and muscle tone, and increase in abdominal fat), depression, cognitive defects and metabolic disturbances (hyperglycemia, altered lipoprotein profile, decreased insulin sensitivity and osteoporosis). 16 Intermittent therapy has been advocated as a measure to reduce morbidity of treatment based on the premise that cycles of ADT followed by re-exposure may delay androgen independence and improve quality of life (QoL).5,17 In a meta-analysis of randomized trials comparing intermittent and continuous ADT, intermittent therapy offered some QoL benefits without negatively impacting patient survival. 18
An increasingly recognized AE of ADT has been the potential for an increased risk of cardiovascular (CV) events. 19 In fact, a meta-analysis of eight observational studies, which together included 491,258 men with prostate cancer, showed that the relative risk (RR) of any type of non-fatal CV disease (CVD) was 1.38 [95% confidence interval (CI) 1.29–1.48] for men receiving LHRH agonists compared with men not treated with ADT, with RRs for non-fatal myocardial infarction (MI) and stroke even higher at 1.57 (95% CI 1.26–1.94) and 1.51 (95% CI 1.24–1.84), respectively. 20 Such is the level of concern regarding cardiac safety with LHRH agonists that in 2010, the US Food and Drug Administration (FDA) required manufacturers of LHRH agonists to add a drug label warning about an increased risk of CVD (i.e. heart attack, sudden cardiac death and stroke) and diabetes in men receiving these drugs for prostate cancer, 21 and guidelines recommend screening for and intervention to prevent/treat CVD in men receiving ADT. 5 Notably, CV events that occur in association with LHRH agonists have been observed soon after ADT initiation, as well as later in the treatment course and in patients with no history of CVD, although the risk is generally increased in patients with pre-existing CVD or CV risk factors. 22 On an encouraging note, however, promising observational and preliminary trial data support a reduced CV risk with GnRH antagonists compared with LHRH agonists.23–26
The most recent addition to alternative forms of ADT has been the development of relugolix, a novel, potent, non-peptide, orally active, small molecule GnRH antagonist. This represents the only oral GnRH antagonist developed and recently approved (December 2020) by the FDA for the treatment of advanced prostate cancer.27–29 Relugolix has been reported to provide the efficacy and potential cardiac safety benefits of GnRH antagonists compared with LHRH agonists in a once-daily oral delivery that is therefore not associated with injection-site reactions, which have limited the uptake of injectable GnRH formulations.15,30
Relugolix clinical development
Relugolix phase I studies
The clinical development program for relugolix included an early phase I clinical study in healthy male volunteers. 31 This randomized, placebo-controlled, single- and multiple-dose, dose-escalation, parallel-group study showed that relugolix is readily absorbed in plasma after single and multiple oral dosing. Relugolix achieved rapid, sustained and reversible testosterone suppression to castrate levels, which was facilitated by the administration of a loading dose. Consistent with the mechanism of action of GnRH antagonists, the fall in testosterone was uniformly associated with prior and/or concurrent profound reductions in LH and FSH. The safety profile for relugolix was in accordance with testosterone suppression. A second phase I trial undertaken in Japanese patients with non-metastatic prostate cancer showed pharmacokinetic, pharmacodynamic and safety data consistent with those in normal healthy volunteers, as well as preliminary evidence of clinical efficacy. 32 These data supported daily doses of relugolix ⩾80 mg, with a single-loading dose on day 1, for phase II clinical testing in men with prostate cancer.
Relugolix phase II studies
Two phase II relugolix studies were undertaken in men with prostate cancer (Figure 2). The C27002 (NCT02083185) randomized, open-label, parallel group study enrolled men with advanced prostate cancer (biochemical relapse, newly diagnosed or advanced localized disease unsuitable for immediate curative intent) and evaluated two dose levels of relugolix compared with leuprolide.33,34 The C27003 (NCT02135445) randomized, open-label, parallel group study in men with intermediate-risk, localized prostate cancer requiring neoadjuvant and adjuvant ADT with external beam radiotherapy (EBRT) compared relugolix with degarelix.35,36
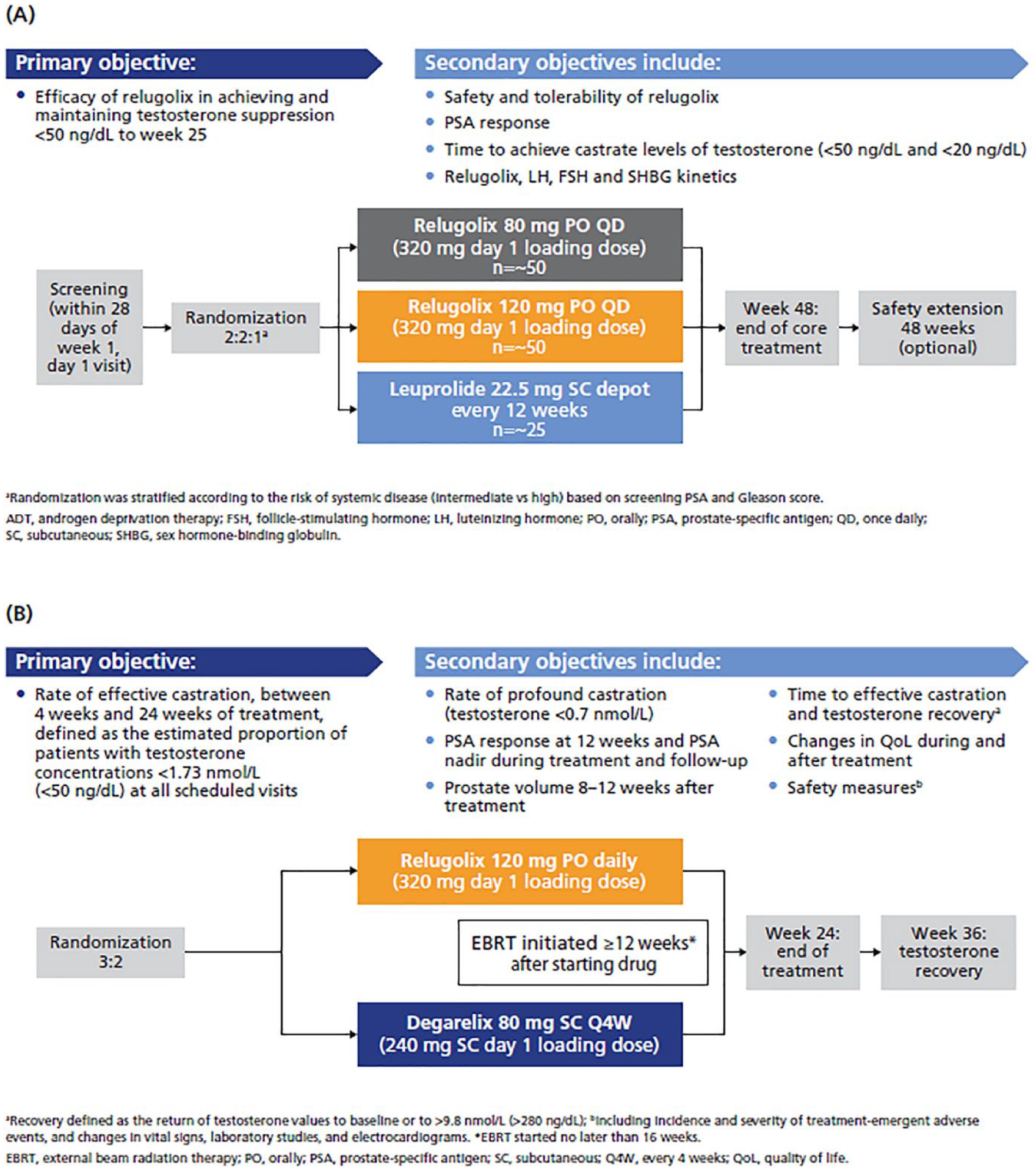
Study designs for C27002 (A) and C27003 (B).
In the C27002 study, 134 patients were randomized to 80 mg (n = 56) or 120 mg (n = 54) once-daily, oral relugolix with a loading dose of 320 mg on day 1, or leuprolide SC 22.5 mg every 12 weeks (n = 24) for up to 48 weeks. 33 The primary endpoint was testosterone suppression (<50 ng/dL) through week 25, with the lower boundary of the 95% CI for at least one of the relugolix groups exceeding 90%. Relative to the leuprolide group, the relugolix groups had fewer patients without metastases, and higher baseline testosterone and prostate-specific antigen (PSA) levels.
Effective (⩽50 ng/dL) and profound (⩽20 ng/dL) castration were achieved and maintained between weeks 5 and 25 in the majority of patients in all treatment groups, but the lower boundary of the 95% CI did not exceed 90% in either relugolix group (or in the leuprolide group); thus, the primary endpoint was not met. 37 While the castration rates were similar for the two relugolix doses, the 120-mg dose trended toward a higher profound castration rate and was more likely to consistently achieve trough drug concentrations >4 ng/mL. 38 In the leuprolide arm, there was an initial surge (91% increase) in median testosterone levels from baseline within 4 days of drug initiation, with a decrease to 67.4 ng/dL by week 3 and profound castration (⩽20 ng/dL) by week 5. In contrast, testosterone levels in patients treated with relugolix decreased immediately, with median values below castration levels within 4 days of drug treatment and profound castration by week 3. For all groups, median PSA levels steadily declined from baseline through week 25 to week 49; PSA reductions by ⩾50% by week 5 were observed in 75%, 83.3% and 16.7% of patients receiving relugolix 80 mg, 120 mg and leuprolide, respectively. PSA suppression by ⩾90% was not observed in the leuprolide group but was achieved by 12.5% and 5.6% of patients in the relugolix 80-mg and 120-mg groups, respectively. FSH suppression was less consistent across the treatment groups, with greater suppression with relugolix than leuprolide.
The safety profile was similar across the treatment groups and was characterized by events associated with medical castration. Notably, the increase in relugolix daily dose from 80 mg to 120 mg was not associated with a substantial incremental toxicity burden.
Degarelix was utilized as the comparator drug in the C27003 trial to provide a contemporary GnRH antagonist benchmark for relugolix, but the study was neither designed nor powered to make formal statistical comparisons between the two drugs. 36 Patients were randomized to oral relugolix 120 mg once daily, with a loading dose of 320 mg on day 1 (n = 65) or degarelix 80 mg SC every 4 weeks, with a loading dose of 240 mg SC on day 1 (n = 38). In both groups, EBRT commenced 12–16 weeks after ADT was initiated. The primary endpoint was rate of effective castration between weeks 4 and 24. Patient and baseline tumor characteristics were generally well balanced between the treatment groups, and most patients had intermediate-risk disease. Median compliance with study drug, as measured with the electronic patient diary, was >98% in both arms.
At week 24, castration rates were 95% and 89% in the relugolix and degarelix treatment groups, respectively, and profound castration rates were 82% and 68%, respectively. The time to castration was rapid in both groups, at a median of 4 and 3 days in the relugolix and degarelix groups, respectively. Following treatment discontinuation, testosterone recovery was faster with relugolix than degarelix [52% versus 16% of patients with testosterone recovery to baseline or >280 ng/dL (the lower limit of normal range) after 12 weeks]. PSA and prostate volume reductions were similar in the relugolix and degarelix treatment groups, and median PSA levels remained low after treatment discontinuation in both arms.
The toxicity profiles were similar for relugolix and degarelix and were generally consistent with testosterone suppression. Severe (⩾grade 3) AEs were infrequent, and no patients in either group discontinued treatment due to AEs. Except for findings of increased alanine aminotransferase and injection-site erythema in the degarelix compared with relugolix group (13% versus 0% and 11% versus 0%, respectively), the overall AE profile was similar between relugolix and degarelix.
The HERO phase III study of relugolix
The preclinical, phase I and phase II studies informed the design of the HERO study, a large, randomized, global, phase III trial comparing relugolix and the widely used depot-injection LHRH agonist leuprolide in patients with advanced prostate cancer (Figure 3). 30 The primary endpoint was sustained testosterone suppression to castrate levels (<50 ng/dL) from day 29 through week 48. Secondary endpoints included non-inferiority with respect to the primary endpoint (−10% margin); castrate levels (<50 ng/dL) of testosterone on days 4 and 15; profound castrate levels of testosterone (<20 ng/dL) on day 15; PSA response (>50% decrease) on day 15; and FSH level at week 24. Testosterone recovery was evaluated in a subgroup of patients.

HERO study design.
Trial design and patient demographics
Patients were enrolled at 155 centers and randomized 2:1 to receive oral relugolix 120 mg once daily, following a loading dose of 360 mg on day 1, or to a 3-month depot of leuprolide 22.5 mg (or 11.25 mg in Japan, Taiwan and China). Randomization was stratified by geographic region, presence (or not) of metastatic disease and age (⩽75 and >75 years). Eligible patients were required to be a candidate for ⩾1 year of continuous ADT and ineligible for surgical therapy and also included those with evidence of biochemical relapse following local primary intervention with curative intent, newly diagnosed metastatic disease and/or advanced localized disease. Patients who experienced a major CV AE (MACE) within 6 months before study start were excluded; MACE was defined as non-fatal MI, non-fatal stroke, and death from any cause. A total of 934 patients were randomized to receive relugolix (n = 622) or leuprolide (n = 308). The majority (>90%) of patients had at least one of the predefined CV risk factors, but the incidence was similar in the two treatment groups. Treatment adherence was >99% in both groups, and 90.2% and 89.0% of patients in the relugolix and leuprolide treatment groups, respectively, completed the 48-week treatment course.
Efficacy results
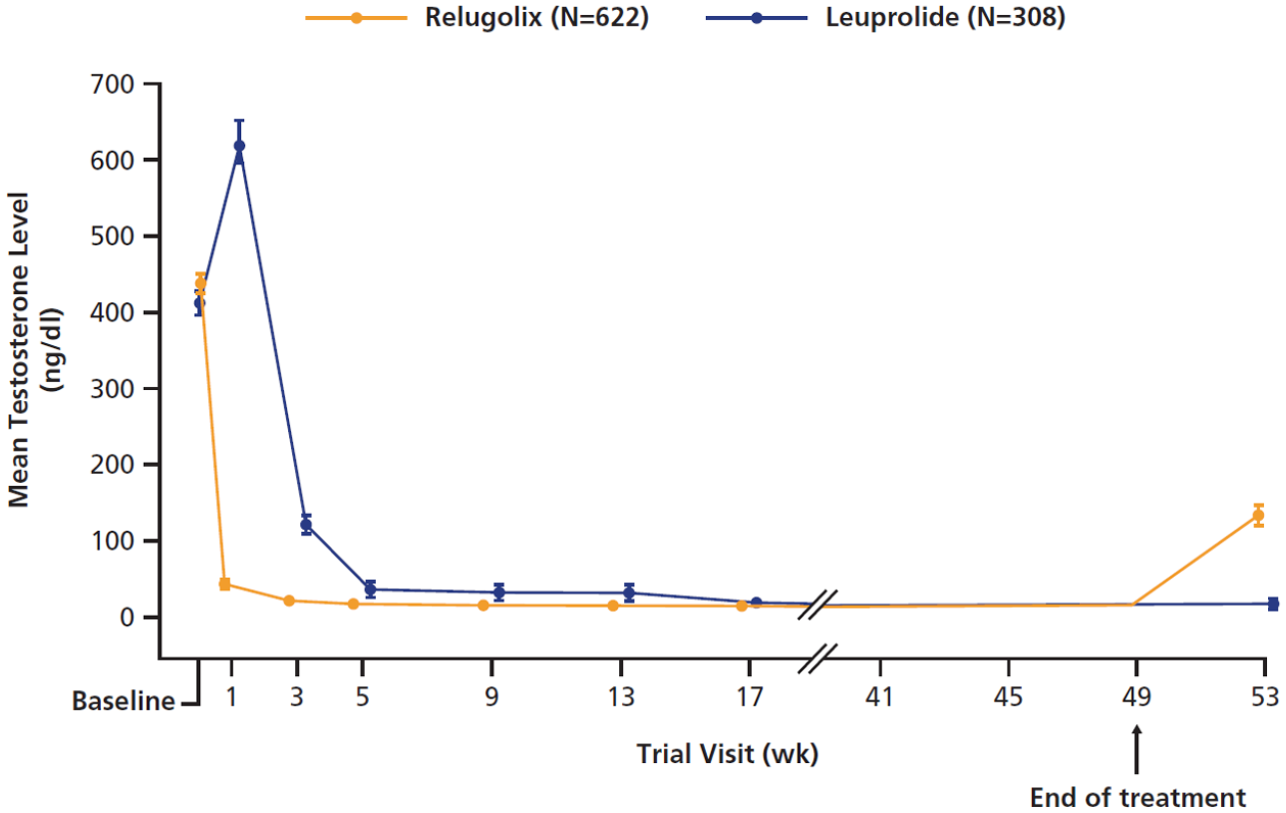
The HERO study met its primary objective, which was that testosterone was suppressed below castrate levels from day 29 through week 48 in 96.7% (95% CI 94.9–97.9) and 88.8% (95% CI 84.6–91.8) of patients in the relugolix and leuprolide treatment groups, respectively. Relugolix was, therefore, non-inferior to leuprolide (between-group difference, 7.9 percentage points; 95% CI 4.1–11.8). Indeed, the lower boundary of the 95% CI for the between-group difference was above zero, which showed the superiority of relugolix over leuprolide (p < 0.001). Relugolix was shown to be non-inferior to leuprolide consistently in subgroups defined by baseline patient and tumor characteristics. Testosterone suppression was faster and more profound with relugolix than leuprolide (Figure 4), with day 4 castration rates of 56% and 0%, respectively (p < 0.001) and day 15 profound castration rates of 78% and 1%, respectively (p < 0.001). Mean testosterone levels on day 4 were 38 mg/dL and 625 mg/dL with relugolix and leuprolide, respectively, which is consistent with the testosterone surge associated with the agonist mechanism of action of leuprolide. Testosterone recovery was faster with relugolix compared with leuprolide (Figure 5), with recovery to ⩾280 ng/dL by day 90 observed in 54% and 3% of patients in the relugolix and leuprolide groups, respectively (p = 0.002). The proportion of patients with a confirmed PSA response at day 15 was also increased with relugolix compared with leuprolide (79.4% versus 19.8%; p < 0.001). Differences were also noted in the FSH suppression profiles for relugolix and leuprolide; FSH suppression was more rapid and profound with relugolix than with leuprolide and increasing levels were noted after day 29 with leuprolide but not relugolix, such that FSH levels were 1.72 IU/L and 5.95 IU/L in the relugolix and leuprolide groups at week 24 (p < 0.001), respectively.
HERO: sustained castration rate.

HERO: mean testosterone levels in the testosterone recovery subgroup (n = 184).
Safety and tolerability
The safety and tolerability profiles were similar in the relugolix and leuprolide groups, with most AEs restricted to grade ⩽2 severity and largely attributable to testosterone suppression; hot flash was the most common AE (50–55% in both groups). Diarrhea was more common with relugolix than leuprolide (12.2% versus 6.8%), but was limited to grade 1 or 2 severity in all cases and did not necessitate treatment discontinuation in any case. Fatal AEs were documented for 1.1% and 2.9% of patients in the relugolix and leuprolide groups, respectively.
After 48 weeks of treatment, the incidence of MACE with relugolix was less than half of that observed with leuprolide [2.9% (95% CI 1.7–4.5) versus 6.2% (95% CI 3.8–9.5)], and Kaplan–Meier estimates showed that the risk of a MACE was 54% lower with relugolix than leuprolide [hazard ratio (HR) 0.46; 95% CI 0.24–0.88; Figure 6]. In patients with a history of MACE, the rates of MACE were 3.6% and 17.8% in patients in the relugolix and leuprolide groups, respectively; this is consistent with a 4.8-fold higher risk of MACE with leuprolide compared with relugolix.

Cumulative incidence of major cardiovascular adverse event at end of week 48 (95% CI).
Clinical relevance of the HERO data
Efficacy and safety
Until the development of GnRH antagonists, ADT choice was limited to LHRH agonists, with variations in injection route (SC, IM injection or SC implant) and frequency of administration (1–12 months). The introduction of GnRH antagonists has expanded the treatment landscape in prostate cancer, but until recently, the only GnRH antagonist available commercially in the US was degarelix, 5 which is available as a 1-month depot SC injection formulation. 14 The results of the HERO study led to the FDA approval of relugolix in December 2020 39 and demonstrate that once-daily, oral administration of relugolix effectively, continuously and reversibly suppresses testosterone to castrate levels. 30
Relugolix represents a novel prostate cancer management strategy, incorporating the mechanistic advantages of GnRH antagonists and the potential benefits of oral administration. Relugolix achieves rapid and sustained castration without the testosterone surge associated with LHRH agonists, thus avoiding the potential clinical consequences of tumor flare and the necessity for concomitant anti-androgen therapy. In the HERO trial, the testosterone suppression observed with relugolix was significantly faster and more profound than with leuprolide. 30 Consistent with the testosterone suppression, the proportion of patients with a PSA response at day 15 was significantly increased with relugolix compared with leuprolide.
In line with its mechanism of action, the rapid testosterone suppression with relugolix was reflected in the rapid and sustained suppression of LH and FSH, with earlier and more profound suppression of LH and FSH observed with relugolix compared with leuprolide. The incomplete FSH suppression observed with leuprolide is a class effect, and although FSH has been identified as having potential oncogenic and metabolic effects, 40 the significance remains to be determined. Testosterone recovery was significantly faster with relugolix than with leuprolide in the HERO study. 30 Additionally, although not powered for statistical significance, testosterone recovery was faster with relugolix than with degarelix in the phase II comparison, which is in keeping with the shorter half-life of relugolix compared with depot degarelix. 36 Intermittent ADT, monitored using PSA levels, is an increasingly attractive management option for patients with prostate cancer as it is associated with improved QoL outcomes. 18 The once-daily oral administration and rapid testosterone suppression and recovery demonstrated with relugolix offer the potential for a flexible administration schedule, which may be of benefit in intermittent therapy, but studies are needed to fully establish optimal utilization in this setting. One potential limitation of rapid testosterone recovery may be that the shorter duration of testosterone suppression with relugolix versus GnRH agonists could impact cancer control if a longer testosterone suppression is actually clinically beneficial. However, this remains to be determined and is currently theoretical. 41 These pharmacodynamic properties of relugolix may also lend support for its use in combination treatment strategies with radiotherapy or cytotoxic therapies, but again, additional studies are required to determine the role for relugolix in combination therapy, with particular attention to potential drug–drug interactions with relugolix.
The safety profile of relugolix in HERO was largely in keeping with that expected for ADT, with hot flash representing the most common AE in both treatment groups. 30 AEs associated with testosterone suppression are unavoidable with ADT, but in HERO, these AEs were generally mild to moderate, with grade 3/4 testosterone suppression-associated events limited to hot flash and fatigue in four and two patients, respectively (all relugolix). It is also encouraging that the rapid testosterone suppression and recovery rates with relugolix may help to reduce drug exposure time and thereby minimize these effects. While diarrhea was increased with relugolix (12% versus 6% of patients in the leuprolide group), as might be expected with an oral agent, it was limited to grade 1/2 severity.
CV safety
There has been a growing body of evidence suggesting that GnRH antagonists have lower CV risk than LHRH agonists in men with prostate cancer, but this is based largely on retrospective analyses, rather than a rigorous assessment of CV risk. Analysis of the World Health Organization database of individual case safety reports of adverse drug reactions revealed that LHRH agonists had a signal both for cardiac events overall (reporting odds ratio [ROR] 1.20) and for specific cardiac events, including MI (ROR 1.76) and heart failure (HF) (ROR 2.02), but GnRH antagonists had no signal for cardiac events overall, although a signal for HF was noted (ROR 1.91). 24 In an Italian retrospective cohort study that had 9785 patients treated with an LHRH agonist (93.6%) or GnRH antagonist (6.4%), the risk of experiencing CV events was significantly lower in patients treated with GnRH antagonists compared with LHRH agonists (HR 0.76, 95% CI 0.60–0.95; p = 0.018); this finding was observed in patients without pre-existing CVD as well as in the overall cohort. 25 A meta-analysis of pooled data from 2328 men in six phase III, prospective, randomized trials comparing LHRH agonists and GnRH antagonists demonstrated that the risk of cardiac events within 1 year of initiating therapy was significantly lower with GnRH antagonists than with LHRH agonists (HR 0.44, 95% CI 0.26–0.74; p = 0.002), but this difference did not manifest in men who had no pre-existing CVD at baseline. 23 A recent, albeit small (N = 80), prospective, randomized trial comparing the 1-month depot GnRH antagonist degarelix and 3-month depot LHRH agonist therapies in patients with advanced prostate cancer and a history of CVD showed that the incidence of MACE was significantly reduced with the GnRH antagonist compared with LHRH agonists (20% versus 3%; p = 0.013). 26
The HERO data represent the largest prospective dataset reporting CV events in patients with prostate cancer treated with LHRH agonists and GnRH antagonists. These data clearly demonstrate an improved cardiac safety profile for relugolix compared with leuprolide. The risk of MACE was approximately 2-fold higher with leuprolide compared with relugolix; this risk differential is even greater (4.8-fold) in patients with a previous history of MACE. The results of the PRONOUNCE trial (NCT02663908) are awaited to further inform our understanding of cardiac safety with LHRH agonists and GnRH antagonists in men with prostate cancer. 42 This ongoing, multicenter, prospective, randomized, open, blinded endpoint trial has been designed to compare the incidence of MACE with the GnRH antagonist degarelix and LHRH agonists in approximately 900 men with prostate cancer. Most men enrolled to date (N = 364) have CV risk factors, including hypertension or dyslipidemia (80%), diabetes mellitus (30%), a previous MI (40%) or previous revascularization (65%).
Potential impact of administration route
While the introduction of GnRH antagonists was heralded as a significant development in the management of advanced prostate cancer, it is likely that safety concerns such as injection-site and hypersensitivity reactions have limited their uptake in the routine clinical setting.12,15 As an oral therapy, relugolix is not associated with injection-related AEs nor indeed with the need for clinic visits for injections. While it is likely that patient preference is for an oral compared with intravenous therapy for prostate cancer, 43 there may be concerns among healthcare practitioners about the potential for poor compliance with oral anticancer therapies, particularly with drugs with a short half-life requiring daily administration, such as relugolix; poor relugolix compliance has the potential to lead to rising testosterone levels and reduced efficacy. While treatment adherence in the HERO trial was notable (>99% in both treatment groups), adherence achieved in a clinical trial with frequent compliance monitoring may not reflect routine clinical practice. There are, however, a number of reasons for optimism regarding relugolix compliance in the real-world setting, including the simple once-daily administration schedule that lends itself to a regular routine (it has been shown that once-daily administration of oral anticancer therapies is associated with significantly improved adherence compared with two-, three- and four-times daily administration 44 ) and the successful adoption and use of the oral once-daily agents abiraterone acetate and enzalutamide, which have demonstrated adherence rates >90% in the real-world management of prostate cancer.45,46 It should also be noted that in the real-world setting, LHRH agonist administration is frequently delayed in comparison with pivotal trial schedules. 47
Financial considerations
Use of any novel therapy may be impacted by its cost relative to currently available treatments. However, to date, there are no published data for the cost-effectiveness of relugolix, but a number of cost-effectiveness analyses of degarelix have been undertaken.47–49 Similar analyses for relugolix would be valuable for healthcare resource utilization decisions.
Conclusions
Relugolix is a novel GnRH antagonist with good efficacy and favorable tolerability, which presents an opportunity to expand the choice of prostate cancer ADT and offer patients the convenience of a once-daily oral therapy. Through rapid testosterone suppression and recovery, relugolix has the potential to minimize ADT AEs and lend itself to combination and intermittent therapy strategies and studies are ongoing to determine the potential role for intermittent relugolix schedules. CV safety is a particular concern in men with prostate cancer; CVD is the leading cause of death in this population, 50 and ADT further increases CV risk. 22 Relugolix has the potential to improve the cardiac safety of ADT and its recent approval represents a valuable addition to the armamentarium for the management of patients with advanced prostate cancer. Work to better understand the reasons behind the differential cardiovascular outcomes with GnRH antagonists and LHRH agonists continues to be a priority.
Footnotes
Acknowledgements
Medical writing support was provided by Tracy McNally and Ify Sargeant of Twist Medical and was funded by Myovant Sciences in compliance with Good Publication Practice 3 ethical guidelines.
Conflict of interest statement
FS reports consulting/advisory role/honoraria from Abbvie, Astellas Pharma, AstraZeneca, Bayer, Janssen Oncology, Myovant, Novartis, Sanofi; Institutional Research Funding from Astellas Pharma, AstraZeneca, Bayer, BMS, Janssen Oncology, Merck, Myovant, Novartis, Pfizer, Sanofi. NDS reports consulting fees from Abbvie, Ambry, Amgen, Astellas, Astra Zeneca, Bayer, BMS, Boston Scientific, Clovis Oncology, Dendreon, Exact Imaging, FerGene, Ferring, Foundation Medicine, Invitae, MDxHealth, Merck, Myovant, Myriad, Nymox, Pfizer, PlatformQ, Sanofi Genzyme, and Tolmar, and speaker’s bureau for Astellas, Bayer, Clovis, Janssen, and Pfizer.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Medical writing support was provided by Tracy McNally and Ify Sargeant of Twist Medical and was funded by Myovant Sciences in compliance with Good Publication Practice 3 ethical guidelines.