
Abstract
Background:
This multicenter study aimed to reveal the genetic spectrum of colorectal cancer (CRC) with deficient mismatch repair (dMMR) and build a screening model for Lynch syndrome (LS).
Methods:
Through the immunohistochemical (IHC) screening of mismatch repair protein results in postoperative CRC patients, 311 dMMR cases, whose germline and somatic variants were detected using the ColonCore panel, were collected. Univariate and multivariate logistic regression analysis was performed on the clinical characteristics of these dMMR individuals, and a clinical nomogram, incorporating statistically significant factors identified using multivariate logistic regression analysis, was constructed to predict the probability of LS. The model was validated externally by an independent cohort.
Results:
In total, 311 CRC patients with IHC dMMR included 95 identified MMR germline variant (LS) cases and 216 cases without pathogenic or likely pathogenic variants in MMR genes (non-Lynch-associated dMMR). Of the 95 individuals, approximately 51.6%, 28.4%, 14.7%, and 5.3% cases carried germline MLH1, MSH2, MSH6, and PMS2 pathogenic or likely pathogenic variants, respectively. A novel nomogram was then built to predict the probability of LS for CRC patients with dMMR intuitively. The receiver operating characteristic (ROC) curve informed that this nomogram-based screening model could identify LS with a higher specificity and sensitivity with an area under curve (AUC) of 0.87 than current screening criteria based on family history. In the external validation cohort, the AUC of the ROC curve reached 0.804, inferring the screening model’s universal applicability. We recommend that dMMR-CRC patients with a probability of LS greater than 0.435 should receive a further germline sequencing.
Conclusion:
This novel screening model based on the clinical characteristic differences between LS and non-Lynch-associated dMMR may assist clinicians to preliminarily screen LS and refer susceptible patients to experienced specialists.
Introduction
Reportedly, approximately 7–15% of colorectal cancers (CRC) are driven by a defective DNA mismatch repair (MMR) system, indicated by microsatellite instability-high (MSI-H) or immunohistochemical (IHC) loss of any of four MMR proteins: MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6), or postmeiotic segregation increased 1 homolog 2 (PMS2).1–3
Notably, there are two main reasons accounting for deficient MMR (dMMR). First, patients diagnosed with Lynch syndrome (LS) carry inherited pathogenic or likely pathogenic (P/LP) variants in any of the five MMR genes, impairing the DNA MMR system. These five MMR genes include MLH1 (MIM: 120436), MSH2 (MIM: 609309), MSH6 (MIM: 600678), PMS2 (MIM: 600259), or Epithelial Cell Adhesion Molecule [EPCAM (MIM: 185535)]. 4 Other dMMR-CRC patients without identified germline pathogenic variants are called non-Lynch-associated dMMR cases, which may be caused by certain epigenetic factors, such as MLH1 silencing by hypermethylation of CpG islands, and loss of heterozygosity, among others.5,6
Many studies have reported the various differences between the Lynch-associated dMMR and non-Lynch-associated dMMR in terms of clinicopathological characteristics, chemosensitivity, and prognosis.7–9 However, there have been few studies attempting to utilize these differences to build a predictive model for CRC patients with dMMR for LS screening. Therefore, we conducted a large-scale, multicenter study involving 15 hospitals from different areas of China. Through ColonCore panel, a next-generation sequencing (NGS)-based panel, we classified dMMR patients into two groups, LS and non-Lynch-associated dMMR, based on whether they carried germline pathogenic MMR gene variants. After collecting, organizing, and analyzing the clinicopathologic differences between the two groups, we generated a novel screening model for LS.
Materials and methods
Patients
We screened the immunohistochemical (IHC) results of MMR proteins (MLH1, MSH2, MSH6, or PMS2) in postoperative CRC patients diagnosed between 1 January 2014 and 1 January 2018, from 15 hospitals across China (Supplemental Table S1), collecting clinical data and pedigree information. The study was approved by the Ethics Committee of the Second Affiliated Hospital of Zhejiang University School of Medicine [approval number: (2017) Ethical Review Research No. 012], and written informed consent was obtained from all patients.
Samples and immunohistochemistry
Tumor tissues with a tumor content ⩾10% and necrotic area ⩽50% were selected, and normal tissues were obtained from negative surgical margins. Meanwhile, white blood cells were used as a normal control when no negative surgical margins were available. DNA was extracted for further sequencing, and IHC staining of formalin-fixed paraffin-embedded tumor tissue was performed to examine the expression of the four MMR proteins (MLH1, MSH2, MSH6, and PMS2) following standard protocols. After deparaffinization, antigen repairing and blocking, the paraffin-embedded slides were incubated with primary antibodies against MLH1 (clone ES05, 1:50 dilution, Dako Cytomation, Carpinteria, CA, USA), MSH2 (clone FE11, dilution 1:50; Oncogene Research Products, Boston, MA, USA), MSH6 (clone EP49, dilution 1:150; Dako Cytomation), or PMS2 (clone EP51, dilution 1:50; Dako Cytomation). We then used the interpretation criteria of the College of American Pathologists (CAP) to determine MMR protein expression. If there was any definite tumor cell nuclear staining, this was considered positive. Conversely, when staining of the tumor cell nucleus was not observed but nuclear staining of adjacent normal cells was present, this was considered to indicate loss of expression. Following this, deficient MMR was defined as the total lack of expression of any of the four MMR proteins. The IHC results were assessed by two specialized pathologists independently.
Laboratory methods
Simultaneous detection of MSI status and variants was performed using ColonCore panel, containing 36 genes from 26 September 2016 to 1 June 2017, and 41 genes after 1 June 2017 (Supplemental Table S2). Additionally, this panel included hereditary CRC related genes, such as MMR genes and Adenomatous Polyposis Coli [APC (MIM: 611731)] along with other genes related to carcinogenesis and CRC development. NGS library preparation, capture-based targeted DNA sequencing, as well as MSI status and germline/somatic variant detection have been described in detail in the Supplemental material. 10 Germline variants were identified as variants detected in both tumor and paired normal tissues. Based on the American College of Medical Genetics and Genomics standards and guidelines for sequence variant interpretation, we classified germline variants according to five tiers: pathogenic, likely pathogenic, uncertain significance, likely benign, and benign. 11 Moreover, hereditary cancer was diagnosed if the patient was accompanied by a germline pathogenic or likely pathogenic variant, as confirmed using Sanger sequencing or multiplex ligation-dependent probe amplification (MLPA).
Construction and validation of nomogram
Variable declaration
The clinical information variables are defined as follows: (1) patient-specific variables, including sex, age at CRC diagnosis, and other LS-associated cancers (endometrial, stomach, ovaries, urinary tract, small intestine, pancreas, bile ducts, brain, and sebaceous glands); (2) personal cancer history defined using three levels: patients with only one CRC, patients with multiple CRCs, and patients with other LS-related cancers; (3) family history of cancer, which was grouped to the following three types: no family history, first-degree relatives (FDR) or second-degree relatives (SDR) affected with CRC, and FDR or SDR affected with other LS-associated cancers; and (4) MMR deficiency defined as four subgroups: MLH1 alone or both MLH1 and PMS2 deficiency, MSH2 alone or both MSH2 and MSH6 deficiency, MSH6 alone deficiency, and PMS2 alone deficiency. Age was grouped into seven subgroups per decade.
Statistical analysis
The clinical characteristic differences between LS and non-Lynch-associated dMMR were analyzed using t tests (continuous variables) and χ2 tests (categorical variables). Multivariate logistic regression with adjusted odds ratio (OR) and with 95% confidence interval (CI) was used to identify independent predictors for LS based on personal history, family cancer history, and MMR deficiency status, building a clinical nomogram based on these independent predictors. For nomogram comparison with other LS screening strategies (Amsterdam II criteria, Bethesda criteria, Chinese Lynch syndrome criteria, and selective strategy proposed by Jiang et al.), 12 the screening sensitivity and specificity of each strategy was calculated based on the genetic testing results. The receiver operating characteristic (ROC) curve was then used to evaluate their discrimination abilities. The Hosmer–Lemeshow test and 200 bootstrapping resamples were used for calibration, and decision curve analysis (DCA) was used to assess the net benefit of nomogram-assisted decisions.
External validation cohort
To examine the nomogram’s universal applicability, an independent external validation cohort of 259 CRC patients with IHC dMMR from the Sun Yat-sen University Cancer Center between November 2011 and December 2015 were included. These 259 cases consisted of 93 cases with and 166 cases without germline pathogenic or likely pathogenic MMR gene variants, whose clinical and genetic information has been published. 12 Sufficient data was available for all patients to score all variables involved in the nomogram. ROC curve, Hosmer–Lemeshow test, 200 bootstrapping resamples, and DCA were also used for external validation based on the data from this cohort.
All statistical analysis was performed using R version 3.5.1 (https://www.r-project.org/). Logistic regression was performed using the rms package, which drew a nomogram, while the ROC curve was drawn using pROC. The ggplot2 package was used to draw histograms and bar plots for visualization of results. The heatmap of multiple genomic alterations were drawn using the ComplexHeatmap package. Differences were considered statistically significant when the two-sided p value was < 0.05.
Results
Germline variant distribution of LS in Chinese CRC
Among the 311 enrolled CRC patients with IHC dMMR, 99 (31.8%) patients carried germline pathogenic/likely pathogenic (P/LP) variants, which included 95 patients with germline P/LP variants in MMR genes (LS) and four patients with germline P/LP variants in other genes (three cases with APC variants and one with BRCA1 variant), as shown in Figure 1. Among those 95 patients diagnosed with LS, about 51.6%, 28.4%, 14.7%, and 5.3% cases carried germline MLH1, MSH2, MSH6, and PMS2 P/LP variants, respectively, and none of the EPCAM-related LS variants was diagnosed. The details regarding clinical characteristics and variant information of LS patients are shown in Supplemental Table S3.
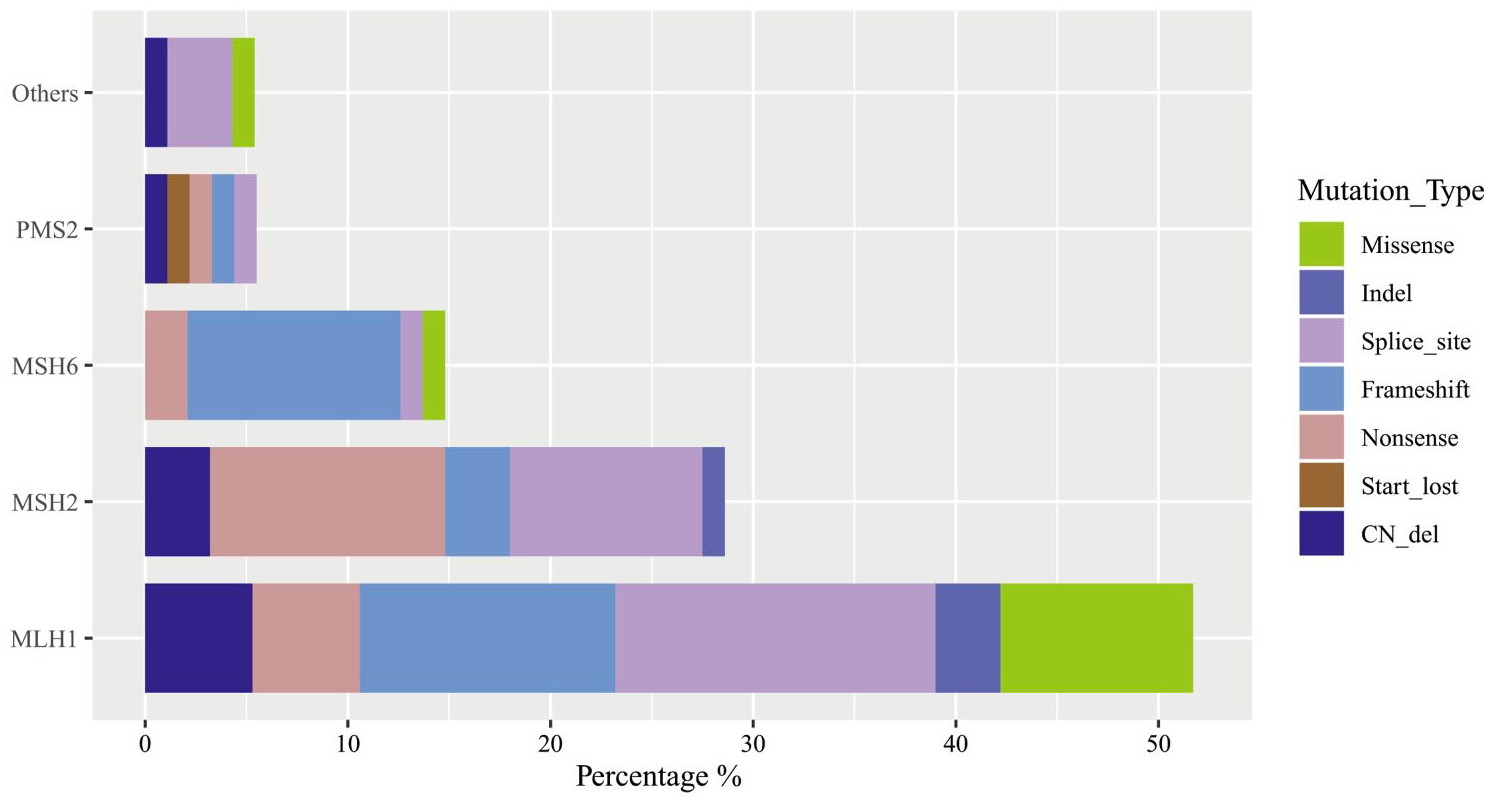
Distribution and types of germline variants in 99 CRC patients carrying germline pathogenic/likely pathogenic variants. Others gene includes APC and BRCA1.
The prevalence of LS ranged widely based on different IHC dMMR patterns. Patients with MSH6 alone deficiency had the highest LS prevalence (60.0%), followed by MSH2 alone or both MSH2 and MSH6 deficiency (41.4%), PMS2 alone deficiency (33.3%), MLH1 alone, or both MLH1 and PMS2 deficiency (23.2%), which have been summarized in Table 1. Interestingly, there were seven (19.4%) cases with PMS2 alone deficiency, which were detected to carry pathogenic/likely pathogenic variants in MLH1 gene but with wild-type PMS2. Similarly, among patients with MSH2 alone or both MSH2 and MSH6 deficiency, two (2.9%) cases were diagnosed with MSH6-related, instead of MSH2-related LS.
Prevalence of LS in CRC patients with different dMMR patterns.
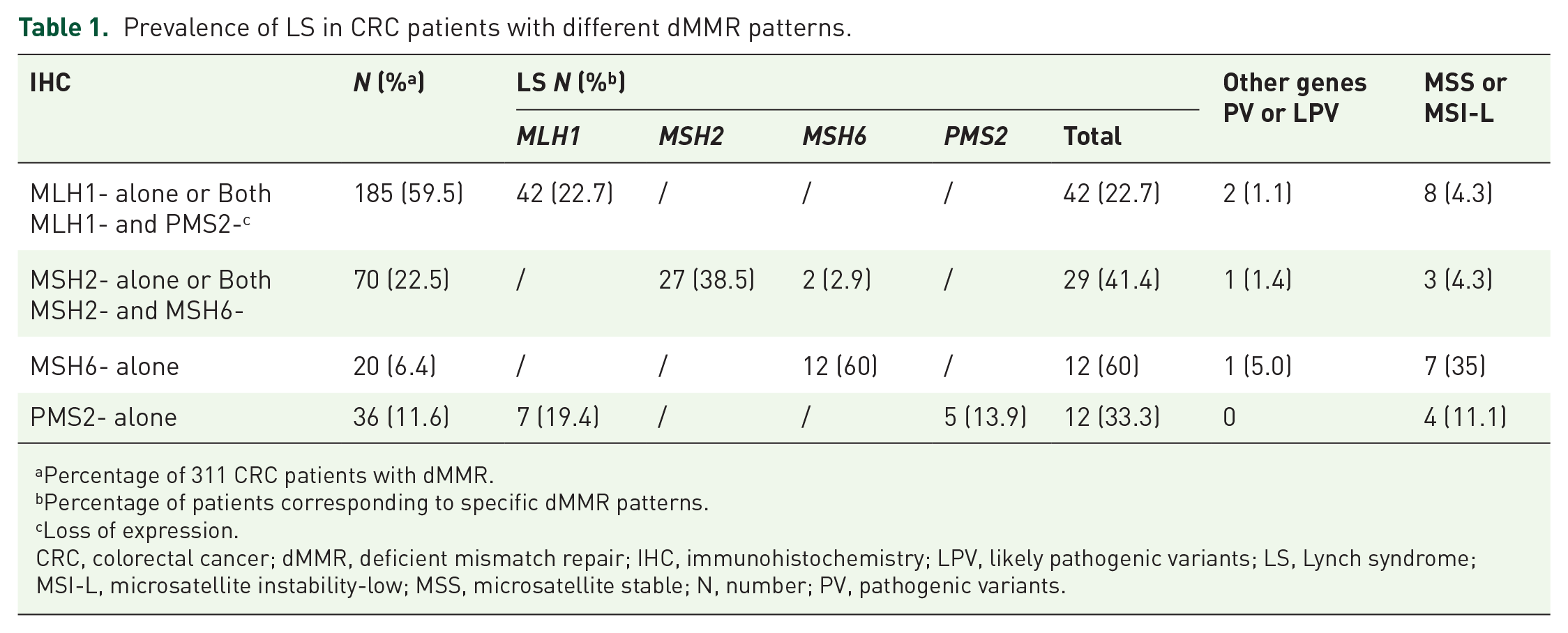
Percentage of 311 CRC patients with dMMR.
Percentage of patients corresponding to specific dMMR patterns.
Loss of expression.
CRC, colorectal cancer; dMMR, deficient mismatch repair; IHC, immunohistochemistry; LPV, likely pathogenic variants; LS, Lynch syndrome; MSI-L, microsatellite instability-low; MSS, microsatellite stable; N, number; PV, pathogenic variants.
Clinicopathologic characteristics and somatic variants of CRC patients with dMMR
Based on whether they carried germline P/LP variants in MMR genes, these 311 CRC patients were divided into two groups: LS (95 cases) and non-Lynch-associated dMMR (216 cases). The clinicopathological characteristics of these two groups are detailed in Table 2. The mean age of patients with LS and non-Lynch-associated dMMR was 44.8 years and 61.2 years, respectively, showing that LS patients were significantly younger than non-Lynch-associated dMMR patients (p < 0.001). Consistent with our expectations, the proportion of LS patients having a family history of CRC was also significantly higher compared with those in the non-Lynch-associated dMMR group (p < 0.001).
Clinicopathological characteristics of CRC patients with dMMR.
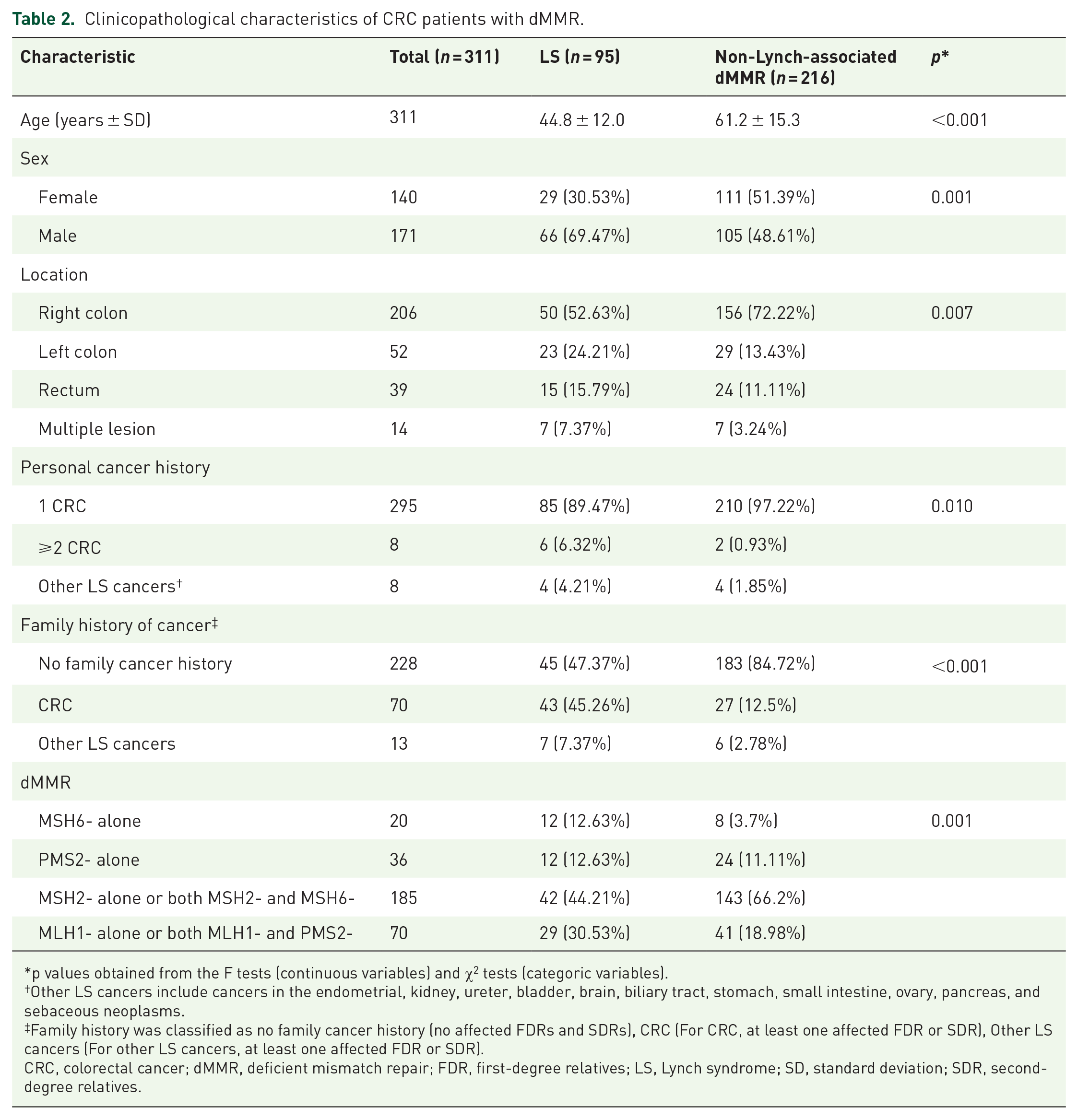
p values obtained from the F tests (continuous variables) and χ2 tests (categoric variables).
Other LS cancers include cancers in the endometrial, kidney, ureter, bladder, brain, biliary tract, stomach, small intestine, ovary, pancreas, and sebaceous neoplasms.
Family history was classified as no family cancer history (no affected FDRs and SDRs), CRC (For CRC, at least one affected FDR or SDR), Other LS cancers (For other LS cancers, at least one affected FDR or SDR).
CRC, colorectal cancer; dMMR, deficient mismatch repair; FDR, first-degree relatives; LS, Lynch syndrome; SD, standard deviation; SDR, second-degree relatives.
Regarding the distinctions of tumor tissue somatic variants between the two groups, the distribution of variant types and the number of variants per sample of LS (Figure 2a, c) and non-Lynch-associated dMMR (Figure 2b, d) are displayed. Furthermore, the top 10 frequently mutated genes from LS and non-Lynch-associated dMMR are shown in Figure 2e. More specifically, except the well-known BRAF V600E mutation, AKT1 gene variants were to occur more frequently in non-Lynch associated dMMR patients (13.0% versus 5.3%, p < 0.05). On the other hand, LS patients were more likely to suffer KRAS-mutant tumors (60.0% versus 38.4%, p < 0.001) and carried a slightly higher mutation rate of APC (69.5% versus 55.6%, p < 0.05). In addition, we found that there was no significant difference between these two groups in the variant frequencies of some key CRC-related genes, such as ERBB2, PIK3CA and TP53 (Figure 2f).

Somatic variant features of the tumors from LS and non-Lynch-associated dMMR. (a, b) Distribution of somatic variant types in LS (a) and non-Lynch-associated dMMR (b). (c, d) Number of variants per sample in LS (c) and non-Lynch-associated dMMR (d) groups. (e) Top 10 frequently mutated genes in tumors from LS (left) and non-Lynch-associated dMMR (right). (f) Mutation frequency of several CRC related key genes, such as APC, ERBB2, and TP53 in LS and non-Lynch-associated dMMR, respectively.
Construction and validation of a novel screening model for LS
Univariate and multivariate analyses of patient characteristics
Due to low prevalence, a screening model with high efficiency and cost-effectiveness may be needed for LS screening. Based on the personal and family history, as well as the various clinicopathological features of these 311 dMMR individuals, we collected several variables, which reached statistical significance in the univariate analysis, including age of cancer diagnoses, sex, tumor location, family cancer history, personal cancer history, and dMMR pattern. Furthermore, all significant variables were entered in a multivariate logistic regression, in which younger age of cancer diagnoses was the most predominant predictive factor (OR: 0.49 per decade; 95% CI: 0.38–0.62), followed by family history, personal cancer history, deficient MMR expression pattern, and sex (Supplemental Table S4).
Construction and validation of nomogram
A nomogram was built by incorporating statistically significant factors identified using multivariate logistic regression to predict the LS probability (Figure 3a). A vertical line was drawn from the factor to the point scale to determine its risk score, and these scores were added up to obtain a corresponding probability of LS.
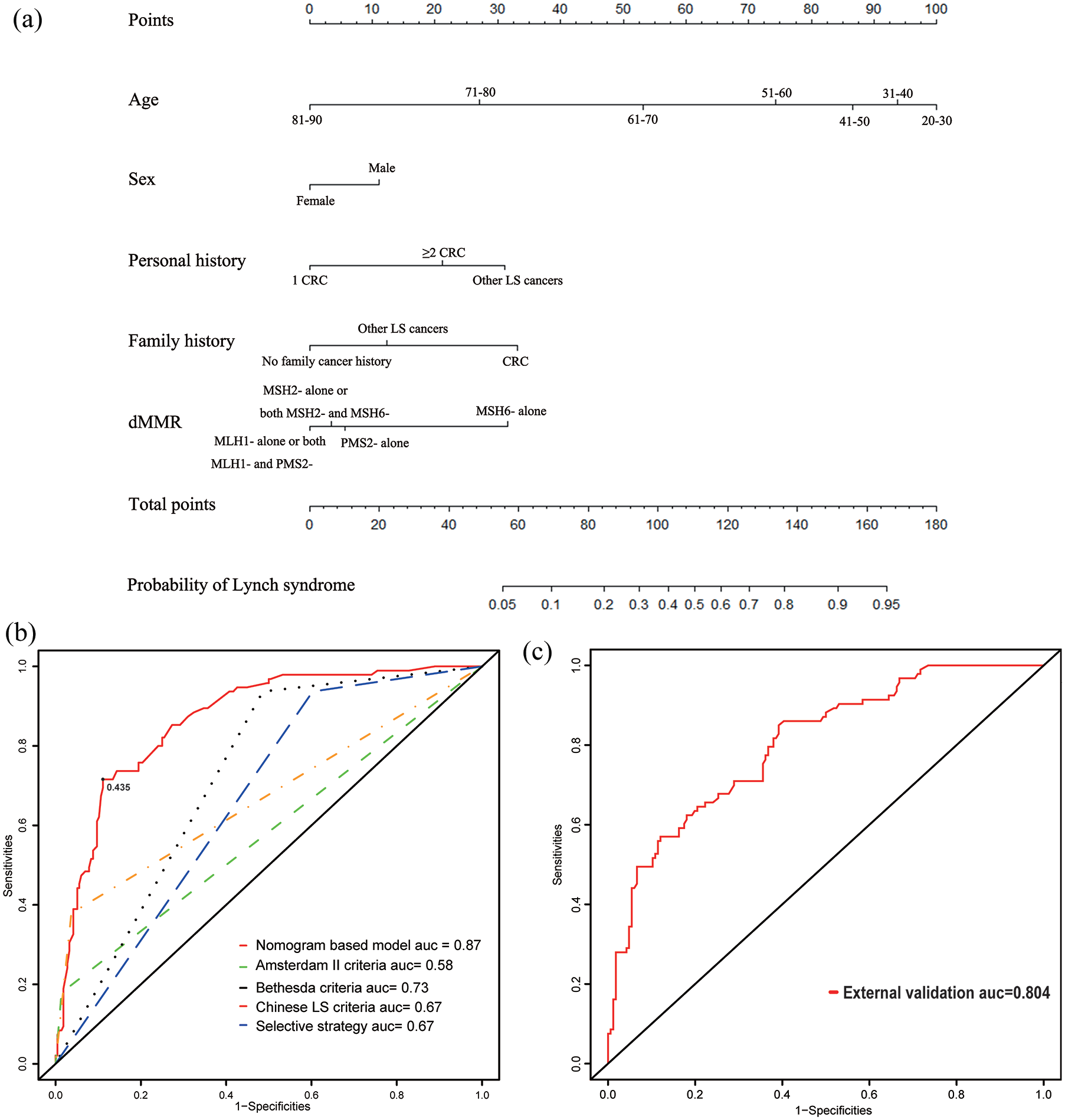
Construction and validation of nomogram to predict the probability of LS. (a) Details of nomogram including the factors of age, gender, personal history, family history, and pattern of dMMR. Other LS-related cancers refer to gastric, endometrial, small bowel, ovarian, and so on. (b) ROC curves comparing the specificity and sensitivity of the nomogram-based model and four current screening criteria to identify LS. The black dot represents the best cut-off value (0.435). (c) ROC curve of the nomogram in the external validation cohort.
The discriminative power of the nomogram-based screening model was quantified by the ROC curves, showing that the nomogram-based screening model is an effective classifier between LS and non-Lynch-associated dMMR, with an AUC of 0.87 (Figure 3b) in the training cohort. Comparing with the ROC curves of Amsterdam II criteria (AUC = 0.58), Bethesda criteria (AUC = 0.73), Chinese LS criteria (AUC = 0.67), as well as the selective strategy (AUC = 0.67) proposed by Jiang et al., 12 this novel screening model could identify LS with higher specificity and sensitivity. Additionally, we suggest that patients with a LS probability >0.435 need to undergo germline sequencing and genetic counseling, as this cut-off value achieves a specificity of 0.889 and a sensitivity of 0.716. To assess the external validation of model performance, we used an independent data set of 259 dMMR patients showing that the AUC of the ROC curve still reached 0.804, which confirms the universal applicability of this screening model (Figure 3c).
The Hosmer–Lemeshow calibration test was not significant for both the training cohort (χ2 = 9.945, p = 0.269) and the external validation cohort (χ2 = 8.158, p = 0.418), indicating a good fit. Internal and external validation with 200 bootstrapping resamples showed relatively good performance for the model (Supplemental Figure S1a, b). Regarding the decision curve analysis (DCA), if the threshold probability in clinical decision was more than 2%, utilization of the nomogram model to screen LS showed a greater advantage than in the assumption that all dMMR patients had LS or that no patients had LS (Supplemental Figure S1c, d).
Discussion
Since 2015, with the rapid development of immunotherapy, the clinical significance of dMMR has no longer been limited as a screening marker for LS. Notably, it has been recommended to routinely perform IHC detection of MMR proteins in tumor tissues of postoperative CRC patients. Thus, we conducted a multicenter study involving 15 hospitals from different areas of China and collected 311 CRC patients with IHC dMMR, including 95 LS cases and 216 non-Lynch-associated dMMR cases. Using univariate and multivariate analysis of the distinctions between LS and non-Lynch-associated dMMR, we generated a novel nomogram for LS screening, showing good discriminatory power in both the training and the external validation cohort.
Nomograms have been accepted as a reliable and alternative tool to predict an individual’s risk of certain clinical events. Through integrating various clinicopathological characteristics, nomograms are able to aid clinical decision making and realize personalized medicine with a user-friendly, convenient, and accurate model. In China, despite the high number of clinicians, only a few of them are well-versed in LS, including its diagnosis, treatment strategies, and family management. Therefore, the vast majority of clinical physicians could utilize this screening model to promptly identify patients with high possibility of LS for referral to genetic counselling by experienced specialists.
Several predictive models for screening LS have been developed, including PREMM5, 13 MMRpredict, 14 and MMRpro. 15 PREMM5, which incorporates PMS2 and EPCAM based on PREMM1,2,6, was developed using data from 18,734 cases using polytomous logistic regression analysis and was validated externally through 1058 individuals. Due to its high sensitivity and specificity, PREMM5 is currently a highly recognized predictive model; however, this model does not incorporate tumor molecular data (MMR or MSI status), and its predictive ability is relatively limited for some patients with weaker phenotypes and who are not affected. MMRpredict, similar to PREMM5, also used logistic regression methodology to analyze 870 CRC subjects diagnosed at under the age of 55 years and was validated by series of patients diagnosed before age 45. Therefore, this model is ideal for predicting the likelihood of patients with young-onset CRC carrying a MLH1, MSH2, or MSH6 variant, but this model does not include information regarding extracolonic cancer history. Unlike MMRpredict and PREMM5, MMRpro estimates the risk of carrying an MMR gene variant based on a Bayesian approach. This model’s unique feature is that it can calculate the probability of carrying a deleterious MMR gene mutation and developing colorectal or endometrial cancer for individuals whose tumor samples are not available. In the aforementioned three prediction models, MMRpro and MMRpredict both incorporate data regarding MMR protein expression or MSI status. In contrast, the prediction model obtained in this study was constructed entirely based on the dMMR population. In China, where family size is declining and IHC results are highly accessible, the nomogram has greater clinical promotion value.
In addition, mainstream predictive models, including the previously mentioned tools, are based mostly on Western population data. However, marked differences in the origin of this disease exist between Eastern and Western populations. Of the major pathogenic LS genes, MSH2 has always been thought to be the most common cause in the Western countries.16,17 On the other hand, several Asian-population studies, including our study, indicated that MLH1 is the most critical causative gene for LS with the range of 40–50% of all LS cases.12,18 In 1998, Yuan et al. gathered 31 Korean families suspected with Lynch syndrome and found that five in seven cases were MLH1 related. 18
During the modeling process, except for age at cancer diagnoses, multivariate analysis showed that a CRC patient carrying other LS-associated cancers, such as endometrial, gastric, and ovarian cancers, was more likely to have LS compared with a CRC patient with multiple occurrences. Similarly, among the four deficient MMR expression patterns, CRC patients with MSH6-alone deficiency are at greatest risk for LS. In addition, although the sex variable did not reach statistical significance (p = 0.075) in the multivariate analysis, we still added it into the nomogram, considering that several researches have reported a relationship between sex and LS.19,20
Moreover, we found that patients with MSH6 alone deficiency had the highest prevalence (60.0%) of LS and had the greatest probability (35%) to be microsatellite stability (MSS) or microsatellite instability-low (MSI-L). This serves as a reminder to pay attention to patients with this variant during LS screening. Additionally, it is not appropriate to select the corresponding gene to be detected based on the deficient dMMR pattern, as a portion of cases with PMS2 alone deficiency are MLH1-related LS and MSH2 alone or both MSH2 and MSH6 deficiency may be from germline pathogenic MSH2 variants.
Despite these findings, there are several limitations in this study. First, the nomogram would be used only for CRC patients with a clear dMMR status since is not suitable for patients whose tumor tissue is not available. Second, although the nomogram has shown high applicability in the external verification cohort, this still needs to be tested and verified in a larger population, especially in Western populations.
In conclusion, through the collection of 311 CRC patients with IHC dMMR, and the analysis of distinctions between LS and non-Lynch-associated dMMR, we created a novel screening strategy for LS with good discriminatory power in both the training and external validation cohort. Due to its convenience and feasibility, this nomogram could be widely popularized in the clinical practice and could potently improve current LS screening in China.
Supplemental Material
sj-docx-1-tam-10.1177_17588359211023290 – Supplemental material for Development and external validation of a novel nomogram for screening Chinese Lynch syndrome: based on a multicenter, population study
Supplemental material, sj-docx-1-tam-10.1177_17588359211023290 for Development and external validation of a novel nomogram for screening Chinese Lynch syndrome: based on a multicenter, population study by Mengyuan Yang, Dan Li, Wu Jiang, Lizhen Zhu, Haixing Ju, Yan Sun, Yuqiang Shan, Chunkang Yang, Jian Dong, Lin Wang, Baoping Wu, Meng Qiu, Xianli Yin, Xicheng Wang, Bin Xiong, Wei Yan, Tao Liu, Chenglin Liu, Xinru Mao, Kefeng Ding, Suzhan Zhang, Shu Zheng, Dong Xu, Peirong Ding and Ying Yuan in Therapeutic Advances in Medical Oncology
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Key R&D Program of China (2018YFC1312100 to Ying Yuan, 2017YFC0908200 to Kefeng Ding); and the National Natural Science Foundation of China (81872481 to Ying Yuan, 81773181 to Dong Xu). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.