
Abstract
The medical management of advanced gastrointestinal stromal tumors (GIST) has improved with the development of tyrosine kinase inhibitors (TKIs) targeting KIT and PDGFRA mutations. However, approximately 5–10% of GIST lack KIT and PDGFRA mutations, and about a half are deficient in succinate dehydrogenase (SDH) that promotes carcinogenesis by the cytoplasmic accumulation of succinate. This rare group of GIST primarily occurs in the younger patients than other subtypes, and is frequently associated with hereditary syndromes. The role of TKIs in patients with SDH-deficient GIST is controversial, with conflicting results; thus, there is an urgent need to uncover the disease mechanisms, treatment patterns, and responses to systemic therapy among these patients. Here, based on an extensive literature search, we have provided a rigorous overview of the current evidence on the medical treatment of SDH-deficient GIST.
Introduction
Approximately 10–15% of adult gastrointestinal stromal tumors (GIST) and almost all pediatric GIST do not harbor mutations in KIT or platelet-derived growth factor receptor alpha (PDGFRA), widely defined as KIT/PDGFRA wild-type (WT) GIST.1,2 The KIT/PDGFRA WT GIST are found to be a rather heterogeneous group of diseases than a single entity, with several different molecular alterations. 3 Furthermore, the KIT/PDGFRA WT GIST can be classified into two main subtypes: succinate dehydrogenase (SDH)-competent and SDH-deficient, according to the SDH immunohistochemical (IHC) status. 4 Moreover, alterations in the RAS signaling pathway (BRAF V600E, or NF1 mutations), neurotrophic tyrosine kinase (NTRK) pathway, and fibroblast growth factor receptor (FGFR) signaling pathway are found in the SDH-competent group.3,4
The SDH-deficient GIST, accounting for approximately 20–40% of all KIT/PDGFRA WT GIST, are thus defined because of loss of expression of SDHB, frequently due to germline and/or somatic loss-of-function mutations in any of the four SDH subunits (A, B, C, or D).4–6 In particular, germline mutations in SDHA occur in approximately 30% of the SDH-deficient GIST, whereas those in SDHB, SDHC, and SDHD occur in only 20–30% of cases.7,8 The remaining 50% of the SDH-deficient GIST lack SDHx mutations, but show hypermethylation of the SDHC promoter. 9 SDH-deficient GIST harboring SDHx germline mutations frequently occur in the setting of hereditary GIST-paraganglioma syndrome, also known as Carney–Stratakis syndrome. 10 This is a rare familial condition inherited as an autosomal dominant trait with incomplete penetrance, characterized by multifocal paragangliomas and GIST, differing from Carney Triad condition due to absence of pulmonary chondromas and presence of SDHx germline mutations. 11
SDH-deficient GIST are easily recognized by clinical features, such as primary gastric localization, multifocality, and indolent clinical behavior, despite being metastatic. 12 Furthermore, SDH-deficient GIST share a distinct molecular background characterized by global DNA hypermethylation, 9 a homogeneous gene expression profile characterized by overexpression of insulin-like growth factor 1 receptor (IGF1R),13,14 and gene expression-based commitment to the neural lineage cell fate. 15
Specific and controlled evidence for an effective therapy for SDH-deficient GIST is lacking, although the hypoxic response to inactivation of SDH complex driven by hypoxia-inducible factor (HIF) transcription factor may represent a rationale for increased sensitivity to tyrosine kinase inhibitors (TKIs) with a prominent antiangiogenic mechanism of action. Moreover, genes involved in glucose metabolism, such as glucose transporter 1 (GLUT1), and angiogenesis, such as vascular endothelial growth factor (VEGF) and macrophage colony-stimulating factor (M-CSF), are upregulated in KIT/PDGFRA WT GIST with respect to mutant GIST. 16 Furthermore, the upregulation of VEGF is described in other SDH-deficient tumors, such as paraganglioma (PGL). 17
The introduction of TKIs revolutionized the treatment of GIST because of the impressive and efficacious control of disease with imatinib as the first-line treatment, especially in KIT and PDGFRA TKI-sensitive mutations. Moreover, sunitinib and regorafenib are approved as the second- and third-line treatments in patients with GIST who develop resistance to imatinib. However, the role of TKIs in patients with SDH-deficient GIST remains controversial and has limited and conflicting results. Therefore, this review aimed to collect all data on response to TKIs in SDH-deficient GIST available in the literature, including the impact of SDH deficiency on the molecular biology of this GIST subset.
SDH deficiency and the pseudohypoxic phenotype of SDH-deficient GIST
The SDH complex, or mitochondrial complex II, serves as a bridge between the Krebs cycle and electron transport chain, and is therefore a key complex in the aerobic respiration. 18 It catalyzes the oxidation of succinate to fumarate, while simultaneously supplying electrons to the respiratory chain complex via reduction of ubiquinone to ubiquinol. 19 The SDH complex is a heterotetramer composed of four subunits: the flavoprotein and succinate-binding SDHA, and iron-sulfur protein SDHB are catalytic subunits; while SDHC and SDHD are the membrane-anchoring subunits responsible for ubiquinone binding. 6 Disruption of the SDH complex due to biallelic loss-of-function mutations or epigenetic inactivation triggers accumulation of the “oncometabolite” succinate that is shuttled between the mitochondrial matrix and cytoplasm. As succinate is structurally similar to α-ketoglutarate (α-KG), it inhibits the α-KG-dependent dioxygenases such as JmjC domain-containing histone lysine demethylases (KDM) and ten–eleven translocation (TET) enzymes20–22 (Figure 1). TET family members are α-KG-dependent dioxygenases involved in DNA demethylation that drives the active removal of 5-methylcytosine (5mC) from methylated CpG sites, leading to transcriptional silencing. Therefore, inhibition of the TET enzymes due to accumulation of succinate induces DNA hypermethylation. 23 Moreover, accumulation of succinate also inhibits the activity of prolyl-hydroxylase domain proteins (PHD) that regulate the oxygen-dependent hydroxylation of HIF-α, leading to their polyubiquitination and proteasomal degradation. 24 Hence, inhibition of PHD impairs degradation of HIF-α, leading to the stabilization of HIF-α and activation of hypoxia-induced gene expression.25,26 Finally, stabilization and upregulation of HIF-α activates the hypoxia-associated angiogenic program.
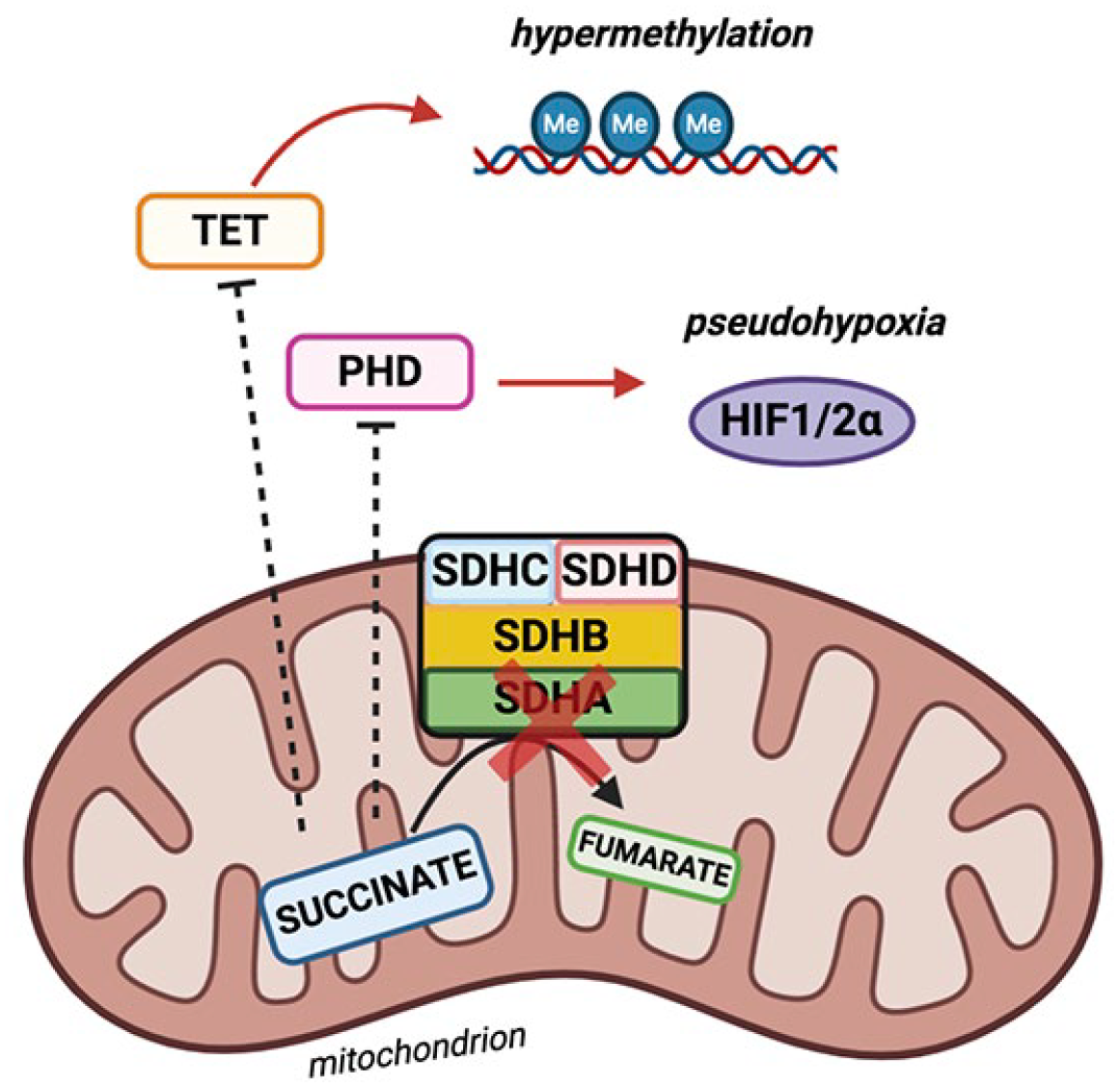
Schematic representing the interplay between hypoxia and SDH-deficient malignancies. Loss-of-function of SDH may lead to accumulation of succinate and production of reactive oxygen species. Additionally, succinate can induce hypoxic response in normoxic conditions, a situation known as pseuodohypoxia.
Overall, SDH-deficient tumors display genomic hypermethylation and activated hypoxic gene response, both derived from the accumulation of succinate and by the subsequent “Warburg effect,” characterized by high glucose consumption and lactate production in cancer cells, despite the presence of oxygen, a feature that is recognized as a hallmark of cancer. 27
The accumulation of succinate due to loss of SDH in chromaffin cells was found to drive DNA hypermethylation, coupled with upregulation of hypoxia-target genes and acquisition of mesenchymal and malignant features; thus, this indirectly suggests that inhibition of HIF-α could serve as a pharmacological target in SDH-deficient tumors. 28 However, these data were primarily derived from in vitro studies on paraganglioma cell models, and not specifically on SDH-deficient GIST cells.21,22,25,26,28,29
Effectiveness of TKIs in SDH-deficient GIST
Due to their rarity, the treatment experience of SDH-deficient GIST is extremely limited, with less data available to date (Table 1). Moreover, most of the data reported in the literature on the activity of TKIs in KIT/PDGFRA wild-type GIST, especially by sunitinib and regorafenib, lack of SDH deficiency characterization, mostly because at the time of publication this molecular characterization was still unknown or unavailable (Table 2).
Available data regarding clinical studies evaluating tyrosine kinase inhibitors in SDH-deficient GIST patients.

1L, first-line; 2L, second-line; 3L+, third- or later-line; CBR, clinical benefit rate; CR, complete response; mPFS, median progression-free survival; MR, mixed response; PFS, progression-free survival; PR, partial response; SD, stable disease; SDH, succinate dehydrogenase.
KIT/PDGFRA wild-type patients GIST patients treated with anti-angiogenic inhibitors.

Sunitinib
Sunitinib is approved as a second-line treatment in patients with imatinib-resistant metastatic GIST as, similar to imatinib, it inhibits the ATP-binding domain in KIT and PDGFRA. 28 Additionally, sunitinib also has an antiangiogenic action, as it inhibits VEGFR, and thus has a broader target activity. 29
In a retrospective study conducted by Boikos et al., 4 87 patients with SDH-deficient GIST were treated with TKIs, and they reported an overall limited efficacy. Moreover, one of 49 patients receiving imatinib achieved partial response (PR) as the best response (2.0%); whereas, in 38 patients with GIST treated with sunitinib, 4 seven patients had an objective response (18.4%), including PR (n = 3), mixed responses (defined as progression at some sites and regression at others; n = 3), and complete response (CR; n = 1). In a retrospective, observational, Chinese study comprising 12 patients with SDH-deficient GIST, four patients (33.3%) developed progressive disease during imatinib treatment; interestingly, all these patients achieved disease control when the systemic treatment was switched to sunitinib. 34
Regorafenib
Regorafenib, the current third-line treatment for metastatic GIST, is another oral, multi-kinase inhibitor that targets several protein kinases, including those involved in angiogenesis (VEGFR1, VEGFR2, VEGFR3, and TIE3).32,43,44 Regorafenib showed a spectrum of activity in KIT/PDGFRA WT GIST in a phase II trial, although only in six patients with SDH-deficient GIST, with PR (n = 2) as the best response (33.3%). 32
Pazopanib
Pazopanib, another KIT, VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR oral inhibitor, has been tested in a phase II study of patients with advanced GIST whose disease progressed on imatinib and sunitinib. 31 A total of 25 patients were enrolled in the trial, including a case of SDH-deficient GIST who showed prolonged disease control after 17 cycles of treatment.
Nilotinib
The second-generation TKI, nilotinib, was designed to overcome resistance to imatinib. Moreover, nilotinib shows activity against BCR-ABL, discoidin domain receptor (DDR), KIT, PDGFR, and colony-stimulating factor receptor-1 (CSF-1R) kinases. 45 In an in vitro setting, nilotinib displayed antitumor activity against imatinib-resistant cell lines, and achieved intracellular concentrations superior than those with imatinib. 46 The clinical experience of nilotinib in SDH-deficient GIST is limited to the report of two young adult patients who achieved remarkable and durable response to 800 mg daily dosage of nilotinib. Additionally, these patients showed stable disease after 42 months and 46 months, respectively, according to the RECIST criteria, with the latter also presenting PR after 9 months according to Choi criteria. 30 Considering the indolent course of SDH-deficient GIST itself, the activity of nilotinib in this subset of patients remains controversial.
Linsitinib
SDH-deficient GISTs are characterized by overexpression of IGF1R.13,14 Thus, the oral IGF-1R TKI linsitinib has been tested in a recent phase II study on adult and pediatric patients with wild-type GIST, including 15 SDH-deficient GIST. 36 No objective responses were detected, but the clinical benefit rate (CBR) and progression-free survival (PFS) at 9 months were 40% and 52%, respectively, suggesting a potential benefit of linsitinib in this patient population.
Vandetanib
Vandetanib, another oral epidermal growth factor receptor (EGFR), VEGFR, and RET inhibitor, has been evaluated in a phase II study of nine pediatric and adult patients with SDH-deficient GIST; however, no CR or PR were observed, and osnly two cases with stable disease were observed, suggesting that vandetanib is not active in this patient population. 47
Discussion
It remains doubtful whether patients with SDH-deficient GIST may benefit from available TKIs, although absence of mutations in KIT or PDGFRA should discourage the administration of imatinib. However, evaluation of the efficacy of TKIs in this patient population is complex. Furthermore, the rarity of this molecular subtype is a potential limit for any prospective trial that could inevitably suffer from a slow accrual, as witnessed in a phase II REGISTRI trial (NCT02638766), a single-arm, multicenter clinical trial aimed to evaluate the efficacy of regorafenib in the first-line setting for metastatic and/or unresectable KIT/PDGFRA WT GIST. 48 Moreover, as the REGISTRI trial focuses on KIT/PDGFRA wild-type GISTs and is not dedicated only to SDH-deficient GIST, the effectiveness data will be stratified according to all different molecular subfamilies of the KIT/PDGFRA wild-type study population. A trial with rogaratinib, a novel pan-FGFR inhibitor, is ongoing, specifically in patients with SDH-deficient GIST who have received at least one standard therapy (NCT04595747). 49 The trial has been opened recently, and no information on the enrollment trend is available yet.
The retrospective evaluation attempted with this systematic review is hampered by the fact that in the past, a vast majority of SDH-deficient GIST were described as KIT/PDGFRA wild-type GIST. Additionally, other preliminary evidence of the clinical benefit of sunitinib in the pediatric and young adult patients with GIST populations was reported, but these data were not evaluated in patients with SDH deficiency as the diagnoses were made a considerable time before. 38 Moreover, variability in genetic determinants of SDH deficiency could also hamper the assessment of definitive conclusions for efficacy of TKI in SDH-deficient GIST. Furthermore, defective function of SDH could be due to different genetic hits, such as truncating or missense mutations affecting different subunits, or promoter hypermethylation; however, no definitive data are currently available on the effects of these variable pathogenetic events on the phenotypic traits deriving from SDH deficiency, such as succinate accumulation, pseudohypoxia, enhanced glycolysis, or hypermethylator phenotype.
To address the issue of limited patient data for this rare GIST subtype, which frequently remains undiagnosed, the Life Raft Group (LRG)—an international nonprofit medical research and advocacy organization—developed an SDH category system to identify patients with potential SDH-deficient GIST and presented results of a retrospective study on patient-reported treatment responses to TKIs and outcomes. 35 With the limitation of a self-reported analysis, a higher percentage of patients who received second-line sunitinib reported tumor shrinkage (36%) than those who received first-line imatinib (14.6%). Moreover, a superior PFS across treatment regimens in patients with Known/Likely SDH-deficient GIST was observed, which could be related to both the greater activity of sunitinib and regorafenib in this cohort of patients and to the indolent behavior of SDH-deficient GIST.
Second, as stated by Boikos et al. 4 due to the frequent indolent behavior of SDH-deficient GIST, stable disease is difficult to interpret as this finding is primarily observed in untreated patients. Therefore, a treatment-related stable disease should be reliable only in patients who have a documented progressive disease during treatment with imatinib or a watchful waiting program. This should be considered as a relevant selection parameter for any clinical prospective trial with this GIST molecular subtype.
Third, in addition to the pseudohypoxic phenotype, SDH-deficient GIST are characterized by global DNA hypermethylation that represents an alternative epigenetic mechanism due to absence of SDH complex not driven by SDHx mutations.50,51 Moreover, the hypermethylation status correlates with aberrant expression of FGF4, by disrupting the binding of CTCF at DNA regions located on the boundaries of the FGF3/FGF4 locus. 52 This FGF4 autocrine loop may act as a surrogate to the KIT/PDGFRA alterations to promote tumorigenesis and growth in SDH-deficient GIST. This feature may explain the greater sensitivity to regorafenib or pazopanib in SDH-deficient GIST, as both also target the FGF/FGFR signaling pathway. Additionally, it was recently found that FGFR1 and FGFR2, as well as FGF4, FGF2, FGF7, and FGF10 ligands are highly expressed in SDH-deficient GIST. 53 This may unleash novel potential treatment strategies for this rare subset of GIST, such as selective FGF/FGFR inhibitors. 54 Consistent with this assumption, the effects of rogaratinib, a novel pan-FGFR inhibitor, is currently being explored in a phase II, single-arm study on patients with a sarcoma harboring an alteration in FGFR-1, -2, -3, or -4 and in patients with metastatic SDH-deficient GIST, regardless of FGFR status. 49
Moreover, due to this hypermethylation phenotype, O6-methylguanine-DNA methyltransferase (MGMT) promoter methylation has been recently found to be markedly prevalent in SDH-deficient GIST. 55 This molecular hallmark could be a proof of concept of sensitivity of SDH-deficient GIST to alkylating agents, as already proven in glioblastoma and neuroendocrine tumors,56,57 and it has also been observed in SDH-mutant paraganglioma and pheochromocytoma. 58 Thus, based on this assumption, a phase II trial of temozolomide is ongoing (NCT03556384) specifically with patients with metastatic SDH-deficient GIST, and not as in two previous trials conducted in an unselected GIST population.59,60
Finally, the IGF-1R inhibition led to no objective responses, but only a good disease control in the overall KIT/PDGFRA WT GIST cohort, predominantly comprising SDH-deficient GIST; this suggests that IGF-1R may not be a pathogenetic driver in SDH-deficient GIST and that the molecular implication of the receptor/pathway in this subset of patients remains complicated to be defined.9,10,45
Identification of a better molecular landscape in SDH-deficient GIST will possibly provide a wider window of potential therapeutic opportunities for this treatment-orphan GIST subtype. 53
Conclusions
SDH-deficient GIST are a rare molecular subset of GIST, that remain unrecognized, with a clinical behavior ranging from a long-term disease stability to an aggressive and rapidly progressive course. SDH deficiency theoretically confers a greater sensitivity to TKIs with a prominent antiangiogenic mechanism of action, even if the efficacy data are very limited and difficult to extract from available studies. Considering the uniqueness and peculiarity of this disease subtype, the only feasible approach is that all patients with SDH-deficient GIST should refer to specialized centers to allow collection of data and reliable clinical evidence.