
Abstract
Antibody–drug conjugates (ADCs) are designed to manipulate the toxic efficacy of specific chemotherapeutic compounds, employing the high affinity of antibody-mediated delivery so as to drive them selectively to target cancer cells. These immunoconjugates encompass the general tendency towards precision medicine and avert the systemic toxicities of conventional chemotherapy, accomplishing an improved therapeutic index. Cumulative experience acquired from first-generation ADCs offers new perspectives to these promising therapeutic modalities for various hematological and solid cancers and propels their clinical development in a faster-than-ever pace, as indicated by the approval of four novel ADCs during the last year. This paper aims to provide an up-to-date overview of the eight ADCs approved by the US Food and Drug Administration and their current indications in oncological practice. Starting from their bio-pharmaceutical background, we track their clinical evolution, with an emphasis on the pivotal trials that led to their commercial release. Late-stage studies examining these eight ADCs in other-than-approved settings as well as the investigation of potential new candidates are also reviewed. In the close future, more data are expected to expand ADCs’ oncological utility and to further reshape their role in cancer therapeutics.
Introduction
The emergence of antibody–drug conjugates (ADCs) in cancer therapeutics has shifted the treatment landscape from conventional chemotherapy to a more targeted approach. Rather than administering unconjugated cytotoxic drugs that destroy all rapidly dividing cells, ADCs are built on a more tailored design, selectively targeting tumor cells with lower systemic toxicity and improved benefit–risk ratio.1,2 As with systemic chemotherapy, a cytotoxic payload is employed but more fittingly delivered to cancer cells via chemical linkage with a monoclonal antibody (mAb). As is thoroughly explained in the section on the biological mechanisms of ADCs’ action, clinical efficacy is achieved through combining the pharmacokinetic profile and specific binding properties of mAbs with the cytotoxic potency of cell-killing agents.
Historically, the first effort to design an efficient ADC can be traced back to the 1960s, when Mathé et al. published a methotrexate-conjugated mAb exhibiting specific anti-proliferation activity against L1210 leukemia cells.3,4 Over the next 60 years, major bioengineering advances, including the development of humanized or fully human mAbs, their linkage with highly potent cytotoxic molecules and troubleshooting of several limitations, such as immunogenicity, attenuated drug delivery, and suboptimal selectivity, were required in order to upgrade ADCs from a theoretical concept to an anticancer option applicable in everyday clinical practice. However, despite the large number of ADCs in pharmaceutical pipelines, a relatively small proportion has reached phase III trial level, out of which only the following eight have received regulatory approval for hematological/oncological indications: (1) gemtuzumab ozogamicin, (2) brentuximab vedotin, (3) ado-trastuzumab emtansine, (4) inotuzumab ozogamicin, (5) polatuzumab vedotin, (6) enfortumab vedotin, (7) fam-trastuzumab deruxtecan and (8) sacituzumab govitecan.
In anticipation of the results for more than 100 ADCs currently being investigated in clinical trials worldwide, we, hereby, focus on the development of the approved ADCs, beginning from the biological rationale behind their designation and following their milestone clinical outcomes, which supported their authorization by the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). This overview summarizes the published evidence on the oncological implications of approved ADCs, outlines the latest ADC-related cancer research and discusses the main concerns rising from their utilization so far as well as their more realistic prospects in the near future.
Biological behavior and mechanisms of action
ADCs are complex biochemical compounds that consist of three key components: an antibody as a nanoscale carrier, a super-toxic drug potent in subnanomolar concentrations, and a sufficiently stable chemical linker that holds them together (Figure 1). The efficacy of an ADC depends on the successful role of each constituent separately while its mechanism of action is based on a sequence of extra- and intracellular events. After their intravenous administration, ADCs circulate in the bloodstream as inactive assemblies and bind to the target antigen on the surface of cancerous cells via the Fab fragments of their antibody component. The optimal mAb component should be characterized by minimal immunogenicity, sufficient antigen specificity and affinity, as well as a long-circulating half-life (as determined by the immune interactions of the constant Fc fragment); while the ideal target antigen should have largely consistent expression on cancer cells (such as lineage-specific markers of CD22, CD30, and CD33), it should have negligible or no expression at all on normal cells to limit off-tumor toxicity. The internalization of the ADC–antigen complex is mediated by receptor-based endocytosis. The intracellular trafficking of the formed endosome containing the prodrug culminates in its fusion with an activated lysosome. In the proteolytic and acidic lysosome microenvironment, the linker, whether or not cleavable, is rapidly degraded and the harbored payload is released to exert its cytotoxic activity. Depending on their intracellular targets, ADC payloads can induce either DNA damage (e.g. DNA double-strand breakers, DNA alkylators and DNA intercalators), or microtubulin disruption (e.g. maytansines and auristatins). 5 Interestingly, some cancer cells, due to de novo or acquired drug resistance, have upregulated efflux pumps and can excrete the cytotoxic payload out of the ADC-targeted cells. 6 However, the liberated payload might also be able to diffuse through the phospholipid bilayer, penetrate and kill neighboring tumor cells, thus evoking a phenomenon known as bystander effect. 7 To a certain degree, this bystander phenomenon can be exploited to overcome tumor heterogeneity, allowing for an off-target albeit in-tumor spread of the cytotoxic agent, regardless of the presence of the target antigen. 8 In breast cancer, trastuzumab deruxtecan serves as a powerful example of how the bystander killing mechanism can be translated into significant therapeutic benefit.

ADC mechanism of action.
Briefly, an ADC acts as a “Trojan horse” for the tumor cell, employing a molecular vehicle to facilitate penetration into the intratumoral territory. This masking protects the payload from elimination while in blood circulation and the tethered drug is discharged from the mAb scaffold to directly elicit its cytotoxic effect upon uptake by targeted cancer cells.
Oncological implications of ADCs
Profound knowledge of these tripartite immunoconjugates in clinical practice is provided by the large number of completed trials on approved ADCs and is expected to further culminate as a result of ongoing studies in the field. In the section below, we elaborate on the main findings during the clinical testing of approved ADCs. Table 1 summarizes all approved ADCs for oncological indications. Table 2 presents several representative trials examining these approved ADCs in distinct malignant indications, combinations, and doses, whereas Table 3 describes some late-stage trials investigating nonapproved ADCs in cancer settings.
Approved ADCs with their oncological indications and their characteristics.
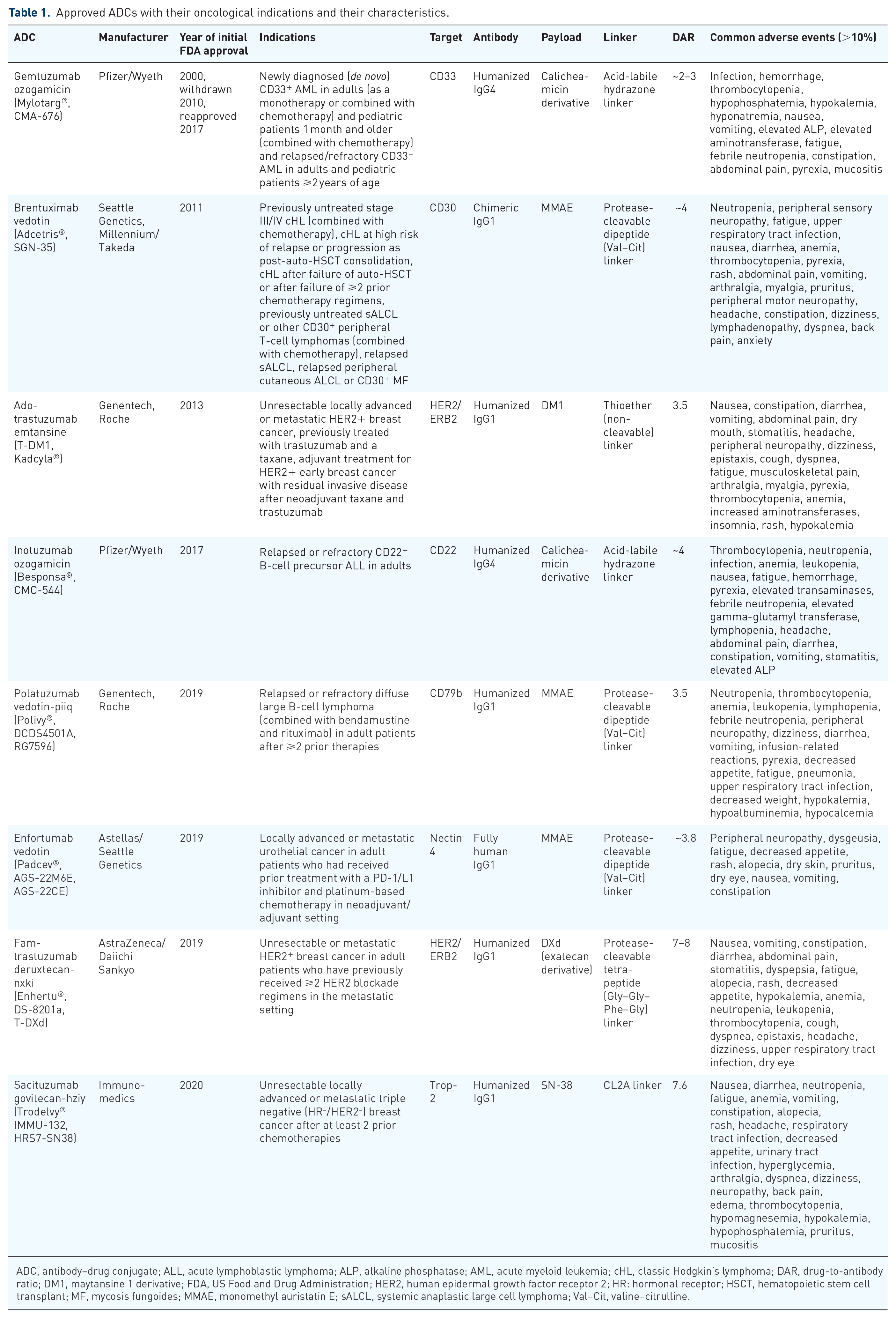
ADC, antibody–drug conjugate; ALL, acute lymphoblastic lymphoma; ALP, alkaline phosphatase; AML, acute myeloid leukemia; cHL, classic Hodgkin’s lymphoma; DAR, drug-to-antibody ratio; DM1, maytansine 1 derivative; FDA, US Food and Drug Administration; HER2, human epidermal growth factor receptor 2; HR: hormonal receptor; HSCT, hematopoietic stem cell transplant; MF, mycosis fungoides; MMAE, monomethyl auristatin E; sALCL, systemic anaplastic large cell lymphoma; Val–Cit, valine–citrulline.
Summary of recruiting phase III trials examining approved ADCs.

ADC, antibody–drug conjugate; AML, acute myeloid leukemia; BV, brentuximab vedotin; DLBCL, diffuse large B-cell lymphoma; DC, daunorubicin, cytarabine; EV, enfortumab vedotin-ejfv; GO, gemtuzumab ozogamicin; HL, Hodgkin’s lymphoma; IM, intramuscular; InO, inotuzumab ozogamicin; IT, intrathecal; IV, intravenous; MRD, minimal residual disease; NCT, ClinicalTrials.gov identifier; PO, per os; PV, polatuzumab vedotin-piiq; R/R, relapsed or refractory; SC, subcutaneously.
Summary of phase III trials investigating nonapproved ADCs.

ADC, antibody–drug conjugate; BM, belantamab mafodotin; DM4, maytansine 4 derivative; HER2, human epidermal growth factor receptor 2; IV, intravenous; MMAF, monomethyl auristatin F; NCT, ClinicalTrials.gov identifier; PO, per os; Q3W, every 3 weeks; Q4W, every 4 weeks; QW, every week; RP2D, recommended phase II dose; RP3D, recommended phase III dose; SC, subcutaneous; seco-DUBA, seco-duocarmycin-hydroxybenzamide-azaindole; VRd, bortezomib, lenalidomide and dexamethasone.
Currently approved ADCs
Gemtuzumab ozogamicin (Mylotarg®, CMA676)
Gemtuzumab ozogamicin (GO) is a CD33-targeting ADC developed for the treatment of CD33+ acute myeloid leukemia (AML). GO involves a semisynthetic derivative of calicheamicin bound via an acid-labile hydrazone linker [4-(4-acetylphenoxy)butanoic acid] to a humanized anti-CD33 IgG4 mAb (hP67.6). 9 Two or three calicheamicin molecules are attached to each IgG4. The therapeutic target antigen, CD33, is a transmembrane glycoprotein belonging to the Siglec family and presents predominantly on myeloid lineages. CD33-expression analysis conducted through standardized flow cytometric assays reported CD33 presence in up to 90% of AML myeloblasts.10,11
Interestingly, this ADC was voluntarily withdrawn and eventually reintroduced after extensive preclinical and clinical testing. GO is currently indicated in combination with three-cycle chemotherapy (daunorubicin and cytarabine) for the treatment of newly diagnosed CD33+ AML in adults, in combination with standard five-cycle chemotherapy in pediatric patients aged 1 month and older and as a monotherapy for relapsed or refractory (R/R) CD33+ AML in patients over 2 years of age. GO can also be used as a monotherapy for adults with newly diagnosed CD33+.
In 2000, the initial accelerated US FDA approval of Mylotarg was based on the results of three open-label single-arm phase II clinical trials in patients with CD33+ AML in first relapse. 12 A total of 142 patients were enrolled and treated with a dosing schema of 9 mg/m2 administered for up to three doses with an interval of 2–4 weeks. Trial outcomes yielded an overall response rate (ORR) of 30%, reflecting an aggregate of 42 patients achieving complete remission (CR) or CR with incomplete platelet recovery (CRp). The median overall survival (OS) was 12.6 months for patients with CR, 11.1 months for patients with CRp, but only 2.9 months for those without CR, leading to a median OS of 5.9 for the entire cohort. 12 Nevertheless, the clinical utility of GO was not confirmed in the post-approval, phase III comparative study S0106 [ClinicalTrials.gov identifier: NCT00085709]. This trial recruited 637 patients (18–60 years old) who were randomized between standard induction chemotherapy with daunorubicin and cytarabine (DC) or standard induction plus ADC treatment with GO. No statistically significant superiority of the combination of GO with DC was detected over the conventional chemotherapy (CR: 69% versus 70%, p = 0.59). 13 These negative results in addition to the higher mortality rate identified in the ADC-containing regimen when compared with the control group (5% versus 1%, p = 0.0062) led Pfizer to voluntarily withdraw GO in June 2010. However, the S0106 study received critiques considering the dosing schedule. More specifically, the high doses of daunorubicin (90 mg/m2) adopted in the control arm have been proven to provide better CR rate and OS when compared with the lower administered doses in the GO+DC regimen. The interesting observation that CD33 binding sites are constantly renewed while surface antigen density is restored in pretreatment levels just 72 h after the ADC delivery led the authors to suggest that concomitant fractioned doses could improve the therapeutic window of Mylotarg. 14 In order to test this hypothesis, another phase III trial, ALFA-0701 [ClinicalTrials.gov identifier: NCT00927498], was conducted, randomizing 271 patients (aged 50–70 years old) with untreated AML who were to receive either standard induction with DC or DC plus GO. In the experimental arm, GO was administered intravenously (IV) 3 mg/m2 on days 1, 4, and 7 during induction and 3 mg/m2 on day 1 in consolidation for patients with remission after induction regimen. As in S0106, CR rates (69.6% versus 70.4%) were similar between the two arms but the median OS was prolonged in ADC-treated group (21.8 versus 27.5 months) without reaching statistical significance. The notably superior finding of this trial was the significantly longer median event-free survival (EFS) in the GO arm compared with the control arm (17.3 versus 9.5 months, p = 0.0002). With regard to safety, drug discontinuation in the experimental arm was mainly due to thrombocytopenia (15.3%) and hepatobiliary-associated adverse events (AEs) (6.1%). Other severe (grade 3–4) AEs included infections (77.9% versus 77.4%), hemorrhage (22.9 versus 9.5%), and veno-occlusive liver disease (VOD) (4.6% versus 1.5%) in the ADC-intensified and conventional induction groups, respectively. In contrast with the S0106 trial, the design of fractioned doses with higher frequency resulted in decreased early mortality rate. 15 Based on these results GO was eventually re-approved by the US FDA in September 2017 (EMA approval on 19 April 2018).
The major body of evidence supporting Mylotarg as monotherapy for AML was provided by two studies, AML-19 [ClinicalTrials.gov identifier: NCT00091234] and MyloFrance-1. The first one, AML-19, was a randomized phase II/III trial that compared efficacy of GO as single agent versus best supportive care in elderly patients (older than 61 years). The main finding was an OS benefit that was achieved with GO (4.9 versus 3.6 months, p = 0.005). Treatment emergent AEs did not vary between the two arms. 16 The second study, MyloFrance-1 was an uncontrolled phase II trial that tested GO in adult patients over 18 years old with AML in first relapse. The fractioned-dose regimen was followed (3 mg/m2 on days 1, 4 and 7) leading to a Complete Response Rate (CRR) of 26%, and to a median OS and relapse-free survival of 8.4 and 11.0 months, respectively. 17
In June 2020, the indications of Mylotarg were extended to include newly diagnosed CD33+ AML in pediatric patients 1 month and older. Approval was supported by the outcomes of the randomized phase III AAML0531 study [ClinicalTrials.gov identifier: NCT00372593]. The primary outcome measures were EFS and OS at 3 years. A total of 1022 patients aged 0–29 years, were enrolled and randomly allocated to receive either standard five-course chemotherapy or chemotherapy plus two doses of GO administered in induction and in intensification course. In the GO arm, EFS was significantly improved (53.1% versus 46.9%, p = 0.04), yet such an improvement was not observed in terms of OS (69.4% versus 65.4). 18 CD33 expression was found to be a major determinant of the clinical benefit observed in all risk groups. More specifically, subjects with low CD33 expression [median mean fluorescence intensity (MFI): 34.61] yielded comparable results regardless of GO administration (EFS 53% versus 58%), whereas patients with higher CD33 expression (median MFI >100.7) showed significant benefit (EFS 53% versus 41%, p = 0.005). 19
Brentuximab vedotin (Adcetris®, SGN-35)
Brentuximab vedotin (BV) comprises a CD30-directed ADC that is approved for the treatment of several CD30-expressing lymphomas, including Hodgkin’s lymphoma (HL), anaplastic large-cell lymphoma (ALCL), and different subtypes of T-cell lymphomas. 20 BV components include a chimeric anti-CD30 IgG1 mAb (cAC10, SGN-30) bonded to the microtubule-disrupting agent monomethyl auristatin E (MMAE) via a protease-cleavable linker [maleimide moiety (mc)–valine–citrulline (Val–Cit)–p-aminobenzyloxycarbonyl (PABC)]. This linker-payload combination is of particular importance as it is used in three of the approved ADCs. The assembly comprises an mc, a Val–Cit dipeptide, a self-immolative PABC spacer, and the antineoplastic agent MMAE.21,22 Approximately four MMAE moieties are attached to each mAb. However, despite the broad use of this linker, its circulatory stability may be suboptimal, as extensive deconjugation and formation of albumin adducts in plasma, possibly via direct transfer, is consistently observed in maleimide-linked ADCs. 23 Recent in vivo data provided evidence of premature release of the payload in rodent serum and identified carboxylesterase 1C as responsible for the extracellular cleavage of Val–Cit-PABC-type linkers. 24 Yet, such an interaction is not observed in humans and its significance remains in preclinical settings.25,26 MMAE is a synthetic dolastatin-10 analog that functions as mitotic inhibitor via tubulin polymerization inhibition, leading to the arrest of mitosis and subsequently apoptosis. 27 With regard to the target molecule, CD30 (TNFRSF8) is a transmembrane glycosylated receptor of the tumor necrosis factor superfamily. Its expression is restricted to a small subset of immune cells, such as activated B- or T-lymphocytes and natural killer cells. 20 CD30 was initially targeted by unconjugated mAbs. MDX-060 and SGN-30 exhibited notable growth-inhibitory properties in HL xenograft models, 28 but the outcomes were not confirmed in the subsequent phase I and II trials.29,30 The minimal activity of SGN-30 as anti-CD30 therapy led to its conjugation with MMAE, thus forming BV.
BV was tested for R/R HL and R/R ALCL in two phase II trials in 2009. In the HL trial [ClinicalTrials.gov identifier: NCT00848926], 102 patients were administered 1.8 mg/kg every 3 weeks, for a median of nine cycles. Tumor regression was noted in 94% of patients; ORR reached 75% with a median duration of response (DoR) of 6.7 months. For the 34% of cases that achieved CR, the DoR was 20.5 months. The spectrum of AEs included peripheral neuropathy (42%), nausea (35%), fatigue (34%), neutropenia (19%), diarrhea (18%), and pyrexia (14%). 31 The same dosing schema was followed in the ALCL trial [ClinicalTrials.gov identifier: NCT00866047] for a median of seven cycles, leading to similar results. Of 58 patients, 50 achieved OR (86%), with a median DoR of 12.6 and 33 achieved CR (57%), with a median DoR of 13.2 months. In general, BV was well tolerated, with a similar toxicity profile to the HL trial. 32 Based on these results, BV received an accelerated approval in August 2011 (EMA approval in October 2012) for patients with HL that had relapsed after autologous stem cell transplantation (ASCT) or had undergone ⩾2 chemotherapy regimens and were not candidates for ASCT, as well as for ALCL after failure of at least one previous chemotherapy. As a post-approval commitment, confirmatory phase III trial, AETHERA [ClinicalTrials.gov identifier: NCT01100502] randomly assigned 329 patients to placebo or BV. 33 After a median follow up of 30 months, the primary endpoint of progression-free survival (PFS) assessed by independent reviewers was remarkably higher in the BV arm compared with the placebo one (median, 42.9 versus 24.1 months, p = 0.0013 and 2-year PFS%, 63% versus 51%). Furthermore, at 5 years, the PFS benefit was sustained (59% versus 41%). 34
In November 2017, based on the results of the phase III ALCANZA trial [ClinicalTrials.gov identifier: NCT01578499], the indications for Adcetris were expanded to include pretreated primary cutaneous ALCL and CD30+ mycosis fungoides (EMA approval in December 2017). 35 This study evaluated the safety and efficacy of Adcetris compared with physician’s other choices (methotrexate or bexarotene) in 128 randomly assigned patients. At a median follow up of 22.9 months, BV proved itself to be superior to conventional therapy in terms of OR duration. More specifically, 36 of 64 patients (56.3%) in the experimental arm achieved an OR lasting ⩾4 months versus 8 of 64 (12.5%, p < 0.0001) in the comparator arm.
In March 2018, BV was further approved for the treatment of previously untreated stage III–IV HL (coadministration with doxorubicin, vinblastine, and dacarbazine), and in November 2018 for previously untreated ALCL and other CD30+ peripheral T-cell lymphomas (coadministration with cyclophosphamide, doxorubicin and prednisone) (EMA approval in February 2019). A phase III study, ECHELON-1 [ClinicalTrials.gov identifier: NCT01712490], randomized 1334 patients to receive six cycles of doxorubicin, vinblastine, and dacarbazine (AVD) plus BV or of standard triplet plus bleomycin (ABVD). In the AVD + BV group, the 2-year modified PFS rate was 82.1% versus 77.2% (p = 0.04) in the ABVD group while the secondary endpoints also favored the AVD + BV regimen. 36 Prolonged follow-up results published in March 2020 confirmed the durability of response observed with AVD + BV. 37 A subsequent double-blind, randomized phase III trial ECHELON-2 [ClinicalTrials.gov identifier: NCT01777152] compared BV in combination with cyclophosphamide, doxorubicin and prednisone (BV + CHP) to cyclophosphamide, doxorubicin, vincristine, and prednisone (CHOP) in 452 patients with previously untreated CD30 + PTCL. Patients were treated with BV + CHP or CHOP for six to eight 21-day cycles. The primary endpoint, PFS, was significantly longer with BV + CHP compared with CHOP (48.2 versus 20.8 months, p = 0.011), while the CRR (68% versus 56%, p = 0.0066) and ORR (83% versus 72%, p = 0.0032) were also in favor of BV + CHP. 38 The toxicity profile was comparable between the two arms and consistent with other studies, including BV. Outside its oncological indications, BV is currently investigated in two phase II clinical trials [ClinicalTrials.gov identifiers: NCT03222492 and NCT03198689], in patients with diffuse cutaneous systemic sclerosis; results are expected in September 2020 and March 2021, respectively.
Ado-trastuzumab emtasine (Kadcyla®, T-DM1)
Ado-trastuzumab emtansine is a humanized anti-HER2 IgG1 mAb (trastuzumab), chemically linked to the antimitotic maytansinoid DM1 via a stable, nonreducible thioether linker [N-succinimidyl-4-(N-maleimidomethyl)cyclohexane-1-carboxylate], bearing a drug-to-antibody ratio (DAR) of ~3.5. HER2 receptor is an already validated target as its amplification/overexpression occurs in ~15–30% of breast cancer (BC). 39 Trastuzumab elicits a multilevel antiproliferative effect upon binding to HER2: prevents the dimerization of receptor, halts the signal transduction of MAPK and PI3K/AKT pathways, induces the Fcγ receptor-mediated Antibody-Dependent Cell-mediated Cytotoxicity (ADCC) and inhibits HER2 ectodomain shedding. In addition to these mAb mechanisms, antitumor activity is further enhanced by the antiproliferative potency of the DM1 payload upon endocytosis. 40 Results from T-DM1 in trastuzumab-refractory and HER2-amplified preclinical models provided adequate evidence to warrant Kadcyla’s entry into clinical testing. 41
In the landmark phase III EMILIA trial [ClinicalTrials.gov identifier: NCT00829166], initiated in February 2009, 991 patients, previously treated with trastuzumab and a taxane, were randomly assigned to T-DM1 or to lapatinib plus capecitabine. In the primary analysis, T-DM1, at the pre-established dosage schedule of 3.6 mg/kg every 3 weeks, conferred a median PFS benefit of 3.2 months (9.6 versus 6.4 months in the control arm, p < 0.001). The subsequent interim analysis demonstrated an additional improvement in OS in the T-DM1-treated group (30.9 versus 25.1 months, p < 0.001). Similarly, secondary endpoints favored the ADC-containing arm, exhibiting higher ORR (43.6% versus 30.8%) and longer DoR (12.6 versus 6.5 months) compared with the control group. 42 This efficacy of T-DM1 remained consistent across distinct patient subgroups. Even in a retrospective analysis of patients with BC and baseline brain metastases, T-DM1 exhibited a clear survival advantage (median OS: 26.8 versus 12.9 months, p = 0.008) but without significant differences in central nervous system progression percentages, PFS, and time-to-symptom presentation. 43 The final OS analysis of EMILIA confirmed the survival prolongation in the T-DM1 arm, even after a treatment crossover of 27% of patients from the control group, according to protocol amendment. 44 Safety analysis showed lower rates of grade ⩾3 AEs in patients treated with T-DM1 (48%) compared with those treated with the capecitabine plus lapatinib combination (60%), with the most frequent Kadcyla-induced toxicities being thrombocytopenia (14%), elevated Aspartate aminotransferase (AST/SGOT) (5%), and anemia (4%). 44 Another phase III trial TH3RESA [ClinicalTrials.gov identifier: NCT01419197] compared T-DM1 with physician’s choice (systemic chemotherapy, hormonal therapy or HER2-directed therapy, as single agents or combinations per local practice) in patients who had prior progression on ⩾2 anti-HER2 regimens and recognized a substantially longer median OS (22.7 versus 15.8 months, respectively, p = 0.0007) for the T-DM1-treated group. 45 Finally, Kadcyla was granted US FDA approval in February 2013 (EMA approval in November 2013) as monotherapy for second- or subsequent-line treatment of patients with HER2+ advanced/metastatic BC, pretreated with trastuzumab and a taxane, separately or in combination.
More recently, in May 2019, Kadcyla received US FDA indication for adjuvant therapy of HER2+ early BC when residual invasive disease remains postoperatively after neoadjuvant therapy with taxane and trastuzumab (EMA approval in November 2019). This adjuvant indication was based on the phase III KATHERINE trial [ClinicalTrials.gov identifier: NCT01772472] which enrolled 1484 patients with HER2+ residual BC and randomized them to T-DM1 (3.6 mg/kg) or trastuzumab (6 mg/kg) IV every 3 weeks, for a total of 14 cycles. At a median follow up of 41.4 months, invasive disease-free survival was significantly higher in the T-DM1 group (p < 0.001) while the risk of recurrence or death was reduced approximately by 50% (12.2% in patients treated with conjugated trastuzumab versus 22.2% in those treated with unconjugated trastuzumab). At 3 years, the rates of patients without invasive disease were 88.3% for T-DM1 and 77.0% for trastuzumab-treated participants. AEs were more frequent with T-DM1 and along the same lines with its previous trials in metastatic setting. 46 Currently, more than 50 ongoing clinical trials examine the therapeutic potential of Kadcyla, not only in BC but also in other solid HER2-expressing tumors, such as lung, bladder, and colorectal cancer.
Inotuzumab ozogamicin (Besponsa®, CMC-544)
Inotuzumab ozogamicin (InO) consists of a humanized anti-CD22 IgG4 mAb (g5/44) conjugated via a hydrazone linker [4-(4′-acetylphenoxy) butanoic acid] to a semi-synthetic calicheamicin derivative (NAc ɣ-calicheamicin DMH). The target-antigen, CD22, is a B-lineage-specific transmembrane glycoprotein that belongs to the sialic-acid-binding immunoglobulin-like lectins. In B-lineage acute lymphoblastic leukemia (ALL), CD22 is expressed at a frequency of 93–100%.47–49 Upon binding to an immunotoxin, CD22 displays rapid internalization in contrast with other routinely expressed antigens, such as CD19. Intracellular conjugate degradation leads to the liberation of the calicheamicin derivative, which binds to the minor grove of the DNA and causes strand scission and cell death. During preclinical testing, the immunoconjugate exhibited notable antitumor potency against CD22+ B-cell lymphomas, as it was found to be four- to eight-fold more cytotoxic than the cell-destructing agent alone. 50
InO was granted US FDA approval in August 2017 (EMA approval in April 2017) as a monotherapy for the treatment of R/R B-cell precursor ALL (BCP-ALL), following the outcomes of the pivotal phase III trial INO-VATE-ALL [ClinicalTrials.gov identifier: NCT01564784], which compared InO to conventional chemotherapy. 51 In this open-label, randomized trial, 326 patients diagnosed with R/R CD22+ Philadelphia chromosome (Ph+ or Ph−) BCP-ALL were enrolled and randomly allocated to receive InO or salvage chemotherapy, according to investigator’s choice, with either FLAG (fludarabine, cytarabine, G-CSF), HIDAC (high dose cytarabine), or cytarabine and mitoxantrone. InO was administered IV at 0.8 mg/m2 on day 1 and at 0.5 mg/m2 on days 8 and 15 of each cycle (C1: 21 days, C2–C6: 28 days). Patients achieving CR with or without complete hematological recovery (CRi, complete remission with incomplete blood count recovery) were subsequently treated with 0.5 mg/m2 on days 1, 8 and 15 of each cycle. Patients in the InO arm demonstrated a significantly higher rate of CR/CRi compared with the control arm (73.8% versus 30.9%, p < 0.0001) and a longer duration of remission (5.4 versus 4.2 months, p = 0.0071). Similarly, survival times were significantly prolonged with InO compared with selected chemotherapy (median PFS: 5.0 versus 1.7 months, p < 0.0001 and median OS: 7.7 versus 6.2 months, p = 0.0105). In addition, when compared with the control group, a greater proportion of patients treated with InO proceeded to ASCT, after CR/CRi, (39.6% versus 10.5%, p < 0.0001). Regarding safety, severe (grade ⩾ 3) AEs were comparable in the experimental and control group (51.8% versus 50.3%), with neutropenia (47% versus 44%) and thrombocytopenia (41% versus 58%) being those most commonly observed. However, permanent treatment discontinuation was more frequent in the InO arm when compared with the chemotherapy arm (18.9% versus 7.7%). Furthermore, in the ADC-treated group, VOD was significantly more frequent (13% versus 2.1%) while any-grade hepatotoxicity was detected in 53% of cases as opposed to 36.45% in the chemotherapy group. 51 The recommended dose of InO for adults at high risk of a thrombotic event is currently evaluated in a phase IV study [ClinicalTrials.gov identifier: NCT03677596] that compares the established dose of 1.8 mg/m2/cycle to the experimental dose of 1.2 mg/m2/cycle. Also, for B-cell non-HL, InO had reached a phase III clinical testing level, but the combination of the ADC plus rituximab (R-InO) failed to achieve better results than those achieved from investigator’s choice treatment, including either rituximab plus bendamustine or rituximab plus gemcitabine (ORR: 41% versus 44% and PFS: 3.9 versus 3.6 months). 52
Polatuzumab vedotin (Polivy®)
In the United States, polatuzumab vedotin-piiq (PV) in combination with bendamustine and rituximab was granted orphan drug designation as third-line or later treatment after recurrence or progression to prior lines, for the treatment of patients with R/R diffuse large B-cell lymphoma (DLBCL) in June 2019 (EMA approval in January 2020). PV is composed of a CD79b-directed, humanized IgG1 monoclonal Ab, covalently conjugated to the antimitotic agent MMAE by a mc–Val–Cit–PABC linker. 53 CD79b, a B-lineage specific transmembrane protein, is expressed in most B-cell malignancies and thus, comprises a viable target antigen for ADC development.53,54 Preclinical studies yielded encouraging findings regarding the efficacy of anti-CD79b–vc–MMAE in diverse non-HL cell lines and across all DLBCL molecular subtypes that met the CD79b-expression threshold prerequisite, while PV also conferred superior outcomes over the standard-of-care R-CHOP regimen in mice xenografts. 53 These data propelled PV into clinical testing.
Its market approval was built on GO29365 [ClinicalTrials.gov identifier: NCT02257567], a phase Ib/II global, open-label, randomized study that assessed the safety and efficacy of PV in combination with chemotherapy and mAbs in R/R follicular lymphoma or DLBCL. In phase Ib, the recommended PV dose was identified at 1.8 mg/kg, while in phase II, PV was administered IV on day 2 of the first cycle and on day 1 of subsequent cycles at a dose of 1.8 mg/kg, combined with bendamustine (B) and either rituximab (R) or obinutuzumab (G) for up to six 21-day cycles. 55 These regimens were well tolerated and provided some durable responses in the noncomparative (PV–BR, PV–BG) safety run-in and the pola-BG dose expansion. In a single-arm phase Ib/II trial, evaluation of the triplet PV–BG in patients with R/R DLBCL who were transplant-ineligible and had limited therapeutic options left, CR rate was 29.6%, while median OS reached 10.8 months. Compared with BR, the PV–BR scheme (40 patients per treatment arm) provided more favorable results in terms of CR (40% versus 17.5%, p = 0.026) and OR rates (62.5% versus 25%), at the end of the study. 55 The updated analysis showed longer DoR (12.7 versus 4.1 months), and longer survival times (median PFS, 7.5 versus 2.0 months, p < 0.001 and median OS, 12.4 versus 4.7 months, p = 0.002) for the PV-containing regimen compared with the control regimen. 56 In addition, the PV + BR regimen also had a higher completion rate compared with BR (46% versus 23%), indicating probably a higher rate of lymphoma progression in the BR arm. For both groups, the most frequent grade 3–4 AEs were cytopenias and infections. Although myelosuppression was more evident in patients treated with PV + BR (neutropenia: 46%, thrombocytopenia: 41% and anemia: 28%) than in those treated with BR (33%, 23% and 18%, respectively), transfusions and severe infections were similar between arms. Lastly, 40% of DLBCL patients from all study parts, treated with PV + BR, developed low-grade, reversible peripheral neuropathy. 56
Given that there are few options available for patients with R/R DLBCL unsuitable for ASCT (including those who experienced ASCT failure) as well as lack of effective bridging for chimeric antigen receptor T-cell therapy, the introduction of PV could cover this therapeutic gap, offering durable clinical responses and apparent survival benefit across the study subgroups and regardless of cell of origin or DEL (MYC/BCL2 double-expressor) biomarker status. 55
Enfortumab vedotin (Padcev®, AGS-22ME, AGS-22CE)
Enfortumab vedotin-ejfv (EV) represents a first-in-class nectin 4-targeting ADC that received US FDA approval on December 2019 for the treatment of patients with locally advanced or metastatic urothelial cancer (Ia/mUC) who have been exposed to anti-PD-1/PD-L1 inhibitors as well as platinum-based chemotherapy in previous lines or in neoadjuvant/adjuvant settings. EV includes a fully human, nectin 4-directed IgG1 mAb attached to the microtubule-disrupting factor MMAE via a mc–Val–Cit-PABC linker. Nectin 4, a transmembrane cell-adhesion protein of the immunoglobulin superfamily, was immunohistochemically detected to be markedly expressed in bladder, breast, pancreatic and lung cancer, but presented in low-to-moderate levels in various normal epithelia. 57 This ADC was found to display a dose-dependent tumor growth inhibition effect across xenograft cancer models, including complete eradication in types of bladder and BCs.
EV was granted an accelerated approval, owing to the results of the EV-201 clinical trial [ClinicalTrials.gov identifier: NCT03219333]. EV-201 is an ongoing phase II, two-cohort study of patients with advanced/metastatic urothelial carcinoma (UC), progressive under platinum-based chemotherapy and immune checkpoint inhibition (ICPI) in cohort 1 or under ICPI alone in cohort 2 (still recruiting). 58 Participants received EV at a dose of 1.25 mg/kg IV on days 1, 8, and 15 of every 4 weeks until disease progression or unacceptable toxicity. At data cutoff, ORR was 44%, with a 12% CRR among responders and median time to response of 1.84 months. Median DoR was 7.6 months and median PFS and OS were estimated as 5.8 and 11.7 months, respectively. Subgroup analyses demonstrated responses across all the prespecified subsets, including the nonresponders to prior anti-PD1/PDL1 (ORR = 41%) and the poor prognostic cases with liver metastases (ORR = 38%), with some degree of tumor regression in most evaluable patients (84%). Most common grade 1–2 treatment-related AEs were a rash (75%) and fatigue (50%), whereas serious toxicities were relatively uncommon (e.g. febrile neutropenia had the highest incidence of 4%). In the majority of cases, treatment dose was reduced (32%) or discontinued (12%) due to peripheral sensory neuropathy (9% and 6%, respectively).
These data encouraged numerous trials to investigate EV for UC in different settings. Among others, the recruiting phase Ib multicohort trial EV-103 [ClinicalTrials.gov identifier: NCT03288545] evaluated EV as monotherapy or combined with chemo- and/or immunotherapy for first- and second-line treatment of advanced/metastatic UC, including cohorts for muscle-invasive UC. In early announced results, EV + pembrolizumab combination demonstrated a remarkable ORR, with response durability that exceeded 70% in 45 patients with metastatic UC, unable to receive cisplatin. 59 Based on these results, US FDA granted a breakthrough therapy designation for first-line EV + pembrolizumab in cisplatin-ineligible patients with advanced/metastatic UC, while the EMA decided a granted waiver in all age groups for all indications of EV.
Fam-trastuzumab deruxtecan (Enhertu®, DS-8201a)
Fam-trastuzumab deruxtecan-nxki comprises trastuzumab, tethered to DXd, a topoisomerase I inhibitor, via an enzymatically cleavable, tetrapeptide-based linker (glycine–glycine–phenylalanine–glycine), with a self-immolative spacer.60,61 Among other available HER2-targeted agents, DS-8201a is distinguished by its broader antitumor potency even against BCs without HER2 amplification and with weak HER2 expression (IHC1+ or IHC2+/ISH−), accounting approximately for 50% of BC cases.60,61 This ADC carries a novel drug-linker system with considerable stability (DAR of ~8) and a novel payload, DXd, an exatecan derivative, with a 10-fold higher topoisomerase I inhibitory potency than SN-38, the active metabolite of irinotecan. These structural elements, in addition to favorable pharmacokinetics of short DXd half-life and the limited systemic payload exposure, assure stable drug delivery and efficacy even in low-HER2-expressing and T-DM1 insensitive tumors.60,62 In co-cultures of HER2(+) and HER2(−) cancerous cells, the drug managed to kill both cell types, developing a sustained bystander effect with high membrane permeability. Hence, DS-8201a can overcome intratumoral heterogeneity, remaining efficient even in cancers with HER2− cell subpopulations. 63 On the contrary, T-DM1 and other anti-HER2 ADCs have lower DAR (ranging between 3 and 4) while their efficacy is limited to highly HER2+ models.60,61 Notably, Enhertu is the first site-specific ADC to receive regulatory approval. Site-specific conjugation can be technically achieved through saturation of the eight interchain cysteine residues available on the Ab by linkage to cytotoxic payloads. This technique generates a well-defined structure as it yields strictly controlled DAR distributions and homogenous ADC populations instead of less-favorable species mixtures. As a result, superior pharmacokinetic properties, greater stability, and enhanced therapeutic index are allowed. 64
Enhertu has been approved for the treatment of adults with unresectable/metastatic HER2+ BC, who have previously received at least two HER2-targeted regimens in metastatic setting. This indication of Enhertu is underpinned by a two-part phase I/II DESTINY-Breast01 trial [ClinicalTrials.gov identifier: NCT03248492]. In this pivotal study, extensively pretreated patients with HER2+ BC, who had progressed on or discontinued T-DM1 due to toxicities or other conditions, were randomized in the first part into three different dosing groups (5.4, 6.4, and 7.4 mg/kg) to evaluate the pharmacokinetics and maximum-tolerated dose of this ADC. 65 In the second, open-label part, 184 patients with a median of six prior lines received the established dose, in the first part, of 5.4 mg/kg. Previous therapeutic lines included T-DM1 and trastuzumab (100%), pertuzumab (65.8%), other anti-HER2 agents (54.3%), hormone therapies (48.9%), and other systemic therapies (99.5%). After a median follow up of 11.1 months with a median treatment duration of 10.0 months, trial data were analyzed, providing an independently confirmed ORR of 60.9% (CR and partial remission rates of 6.0% and 54.9%, respectively), with a median time to response of 1.6 months. A disease control rate (DCR) of 97.3% was reached, the clinical-benefit rate achieved was 76.1%, and outcomes were consistent across most study subgroups. At 6 and 12 months, estimated OS was 93.9% and 86.2%, respectively, while the median OS was not reached at the time of trial publication. 65 The pooled safety analysis of HER2+ BC patients treated with Enhertu estimated that a proportion of 20% had experienced serious treatment-emergent AEs. 66 Most common treatment-emergent AEs were gastrointestinal and hematological events. The discontinuation of ADC was permanent in 9% of patients, mainly because of interstitial lung disease (6%). Treatment-related mortality was evaluated at 4.3% and the most common cause of death was pulmonary toxicity/interstitial lung disease (2.6%).
Interestingly, promising activity outcomes have also been reported in a phase I HER2-low BC subgroup analysis, demonstrating confirmed investigator-assessed ORR and DCR of 44.4% and 83.3%, respectively, and median DoR and PFS of 11.0 months and 8.0 months, respectively. Independently assessed ORR was 37.0%, while median PFS and OS were 11.1 and 29.4 months, respectively. 67 Similar activity evaluation has also been conducted across a HER2+ gastric cancer population, but the favorable preliminary findings published require confirmation in subsequent trials (ORR = 43.2%, DCR = 79.5%, median OS = 12.8 months). 68 Overall, trastuzumab deruxtecan was well tolerated and displayed a manageable safety profile, consistent across subgroups, with encouraging, durable responses, thus verifying the preclinical rationale.
In view of these favorable outcomes, recruiting phase III, trials DESTINY-Breast02, 03 and 04 [ClinicalTrials.gov identifiers: NCT03523585, NCT03529110 and NCT03734029] are conducted to directly compare Enhertu with T-DM1 and physician’s choice in HER2+ high and low BC. Recently, a phase II trial DESTINY-Gastric01 [ClinicalTrials.gov identifier: NCT03329690] has met its primary endpoint as Enhertu achieved a clinically significant improvement in ORR compared with investigator’s choice in HER2-expressing metastatic gastric cancer.
Sacituzumab govitecan-hziy (Trodelvy® IMMU-132, HRS7-SN38)
Sacituzumab govitecan-hziy (SG) is the latest next-generation ADC, which attained accelerated US FDA approval for metastatic triple-negative BC (MTNBC) in April 2020. SG consists of a humanized anti-Trop-2 IgG1k mAb (hRS7) tethered via the CL2A linker to the topoisomerase inhibitor I SN-38. Trop-2 (TACSDT2) is a cell-surface glycoprotein upregulated in more than 80% epithelial cancers and identified as an unfavorable prognostic factor.69,70 In BC, overexpression of Trop-2 was associated with histological grade (p = 0.023), lymph node metastasis (p < 0.001), and distal metastasis (p = 0.004). 71 SN-38 is an active metabolite of irinotecan, approximately 1000-fold more potent than the parental drug. SG is a high-loaded ADC, bearing a DAR of approximately 7.6.
US FDA approval was founded on the results of a phase I/II single-arm study [ClinicalTrials.gov identifier: NCT01631552] testing SG in previously treated patients (over 18 years of age) with advanced epithelial tumors. Amongst enrolled patients, 108 received SG for MTNBC as a third or higher line of treatment. SG was administered IV at 10 mg/kg on days 1 and 8 of 21-day cycles (median 9.6) until unacceptable toxicity or disease progression. With regard to the trial’s primary endpoints, OR was observed in 33.3% of patients, while CR was achieved in 2.8%. The median DoR was 7.7 months. In terms of safety, common AEs included nausea, diarrhea, neutropenia, and anemia (Table 1). Grade 3 events were reported in 66%, with the most frequent being neutropenia (26%), anemia (11%), and hypophosphatemia (9%). Grade 4 events were reported in 19%, the most common being neutropenia (14%). AE-related drug discontinuation occurred in three cases. 72 SG is currently being further investigated for R/R MTNBC after at least two prior chemotherapies in the randomized phase III trial ASCENT [ClinicalTrials.gov identifier: NCT02574455]. A total of 529 patients were assigned 1:1 to receive either SG, according to the abovementioned dosing schedule, or TPC (eribulin, gemcitabine, capecitabine, vinorelbine). The primary endpoint is a 3-year PFS. SG is tested versus the same comparator arm for hormonal receptor positive, EGFR-2− metastatic BC in 400 patients in the phase III trial TROPICS-02 [ClinicalTrials.gov identifier: NCT03901339]. 73 Due to Trop-2 overexpression in a plethora of neoplasms, SG is also studied as a monotherapy or in multidrug regimens, in several ongoing trials. Based on results of phase II trials in small cell lung carcinoma and non-small cell lung carcinoma, SG also gained Fast Tract designation from the US FDA for these conditions. Overall, SG holds great promise in the field of ADCs, as supported by SG data of remarkable preclinical potency in tumors, including ovarian, 74 uterine, 75 and cervical 76 cancers.
Conclusion
ADCs are gaining momentum in the future direction of precision medicine, aiming to set a new paradigm shift in cancer-treatment algorithms. Data from completed clinical trials of the abovementioned ADCs have expanded our understanding on targeted therapy, while simultaneously taking preliminary steps outside the oncological spectrum. ADCs are now entering an era of exponential development and many efforts are made to establish them as standard-of-care options for various malignancies, as suggested by the large number of active clinical trials and the increasing interest of the biopharmaceutical community. Further in-depth research and structural optimization is required to follow the ever-growing amount of target candidates and to render ADCs as a prominent addition to our anticancer arsenal.
Footnotes
Author contributions
CT and PPL reviewed the literature and CT, PPL, HG and DZ wrote the manuscript. All authors edited, read and approved the final manuscript.
Availability of data
Published data supporting this article are included within the reference list. Please contact corresponding author for any further requests or supplementary information.
Conflict of interest statement
HG has received grants and personal fees by Roche, BMS, MSD, Novartis and personal fees by Amgen and Pierre Fabre, outside the submitted work. All other authors declare no conflict of interest.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.