
Abstract
Serum and glucocorticoid-inducible kinase 1 (SGK1) is an AGC kinase that has been reported to be involved in a variety of physiological and pathological processes. Recent evidence has accumulated that SGK1 acts as an essential Akt-independent mediator of phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathway in cancer. SGK1 is overexpressed in several tumors, including prostate cancer, colorectal carcinoma, glioblastoma, breast cancer, and endometrial cancer. The functions of SGK1 include regulating tumor growth, survival, metastasis, autophagy, immunoregulation, calcium (Ca2+) signaling, cancer stem cells, cell cycle, and therapeutic resistance. In this review, we introduce the pleiotropic role of SGK1 in the development and progression of tumors, summarize its downstream targets, and integrate the knowledge provided by preclinical studies that the prospect of SGK1 inhibition as a potential therapeutic approach.
Keywords
Introduction
Serum and glucocorticoid-inducible kinase 1 (SGK1) is a member of the AGC kinase family of serine/threonine kinases, sharing a large homologous sequence and kinase function with the Akt family,1–3 but it is distinct from Akt that SGK1 does not possess a pleckstrin homology (PH) domain to directly interact with phosphatidylinositol 3,4,5 trisphosphate. 4 SGK1 was initially cloned as an immediate-early gene in rat breast tumor cells stimulated by serum and glucocorticoids, and its function is closely related to the phosphorylation of mammalian target of rapamycin (mTOR).1,5,6 The kinase converts into an open conformation for phosphorylation after mTOR-dependent hydrophobic phosphorylation on Ser422, and is fully activated by 3-phosphoinositide-dependent kinase-1 (PDK1).4,7 The consensus phosphorylation site of SGK1 includes serine or threonine residues in the substrate proteins present in an RXRXX(S/T) sequence motif (where X is any amino acid, and R is arginine). 1
SGK1 is expressed ubiquitously in most tissues tested to date, and its subcellular localization is associated with the functional state of the cells. SGK1 enters the nucleus after serum stimulation, but may also be located in the cytosol following activation by hypertonic shock or glucocorticoids. 1 SGK expression is regulated by a wide spectrum of stimuli, including dehydration, saline ingestion, glucocorticoids, insulin, growth factors, and various cellular stresses such as cell swelling, metabolic acidosis, neuronal excitotoxicity, ultraviolet irradiation, DNA damage, and ischemia.1,8–13 SGK1 is involved in regulating multiple ion channels, membrane transporters, cellular enzymes, and transcription factors, as well as in a variety of physiological processes including memory consolidation, reproductive process, cell growth, proliferation, survival, migration, and apoptosis.1,3,14,15 SGK1 has been previously reported in hypertension, diabetes, hypercoagulability, and autoimmune disease.5,16,17 A growing number of recent studies reveal a role for SGK1 in human tumors such as glioblastoma,18–20 breast,21–25 colorectal,26–28 prostate,29–31 thyroid, 32 ovarian, 33 myeloma, 11 and non-small cell lung cancer 34 (Figure 1).

The functions of SGK1. SGK1 is involved in a broad range of processes of human physiology and pathophysiology.
With such extensive studies on SGK1, the understanding of its role in cancer is continuing to improve. In this review, we summarize the functions of SGK1 in tumor growth, survival, metastasis, autophagy, immunoregulation, Ca2+ signaling, cancer stem cells (CSCs), cell cycle, and therapeutic resistance, to clarify the roles of SGK1 cross-talk in different pathways (Figure 2; Table 1).
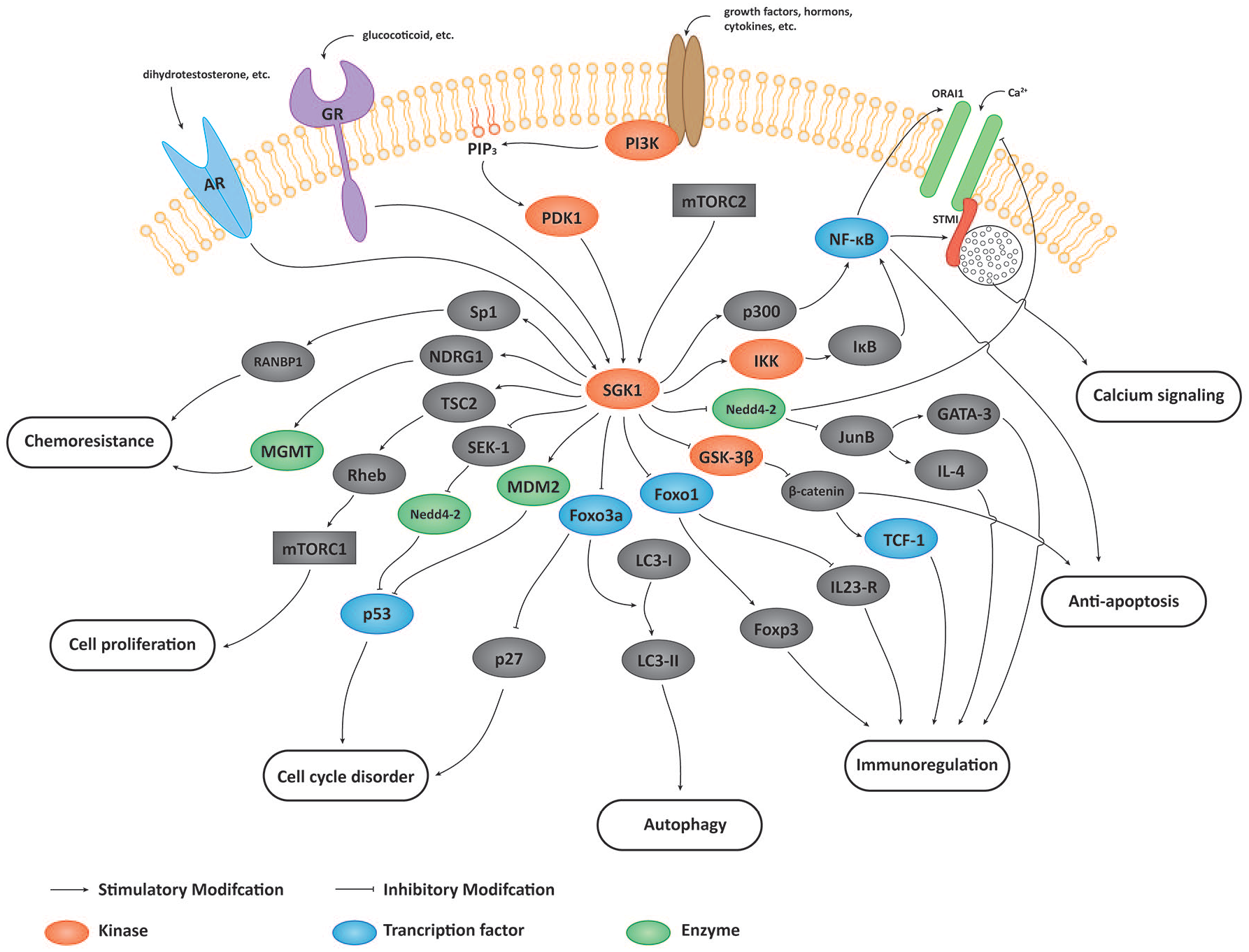
Signaling pathway of SGK1 in oncology. SGK1 regulates tumor growth, survival, metastasis, autophagy, immunoregulation, Ca2+ signaling, CSCs, and the cell cycle through phosphorylating different substrates.
The substrates and regulation mechanisms of SGK1.

SGK1, serum and glucocorticoid-inducible kinase 1.
Multiple factors regulate SGK1 in oncology
SGK1 transcription is stimulated by multiple factors, including hormones, cytokines, stress response, and multiple signaling pathways and physiological processes. 5 In this review, we focus on stimuli with potential roles in the oncological activities of SGK1.
SGK1 is known to be activated by insulin via phosphatidylinositol 3-kinase (PI3K) signaling. SGK1 is phosphorylated by mTOR complex 2 (mTORC2) at Ser422, which offers a docking site for PDK1. PDK1 then fully activates SGK1 by phosphorylating Thr256 in the activation loop of its catalytic domain. 7
In addition to mTORC2 and PDK1, SGK1 activation is also related to other cofactors through transcriptional or posttranslational regulation. During DNA damage, SGK1 is significantly induced in a p53-dependent manner via extracellular signal-regulated kinase 1/2 (ERK1/2). 8 Interleukin-2 (IL-2) also induces SGK1 protein expression, possibly via transcriptional activation and posttranscriptional phosphorylation. 51 Androgen receptor (AR) is a pivotal ligand-activated transcription factor and contributes to the development and progression of prostate cancer. Following androgen stimulation, AR activates the response element motif (5′-CGGACAaaaTGTTCT-3′) at −1159/−1145 region in SGK1 promoter and upregulates SGK1 expression. 30 The glucocorticoid receptor (GR) is another transcription factor, which shares an almost identical consensus DNA-binding motif with AR. 52 The glucocorticoid receptor thus regulates SGK1 expression and enhances cell survival in prostate cancer and triple-negative breast cancer via a similar mechanism.53,54 β2-microglobulin upregulates phosphorylated SGK1/SGK1 level and promotes cell growth and survival in estrogen receptor-negative and HER2-negative breast cancer through the SGK1/Bcl-2 pathway. 55
Functions and mechanisms of SGK1 in oncology
Growth, survival, and metastasis
SGK1 expression is elevated in several tumors, including prostate cancer, colorectal carcinoma, glioblastoma, breast cancer, and endometrial cancer. SGK1 expression is also associated with tumor growth, survival, and metastasis.20,26,49,56,57
The PI3K/Akt/mTOR signaling pathway is abnormally activated in most cancers, and has been considered as a promising therapeutic target. 58 Akt is a well-known classic effector of PI3K-mediated activity and phosphorylates numerous substrates involved in cell growth, proliferation, metabolism, survival, and glucose metabolism. 6 However, growing evidence has recently pointed to the existence of additional effectors of PI3K, that is SGK1 plays a critical role downstream of PI3K.6,23
SGK1 is required for PI3K-activation-related cancer cell proliferation, while the depletion of SGK1 reduces the proliferation and viability of cancer cells in a wide variety of malignancies, including glioblastoma, colon, prostate, thyroid, and endometrial cancers.19,31,32,59,60 Combined targeting of SGK1 and Akt suppresses cell growth more efficiently than inhibiting either PI3K or Akt alone. 32 The activity of mTORC1 is regulated through the tuberous sclerosis complex (TSC)/Ras homolog enriched in brain (RHEB)/mTORC1 axis. SGK1 maintains the activity of mTORC1 by phosphorylating and inhibiting its negative regulator TSC2. 39
In addition to mTOR-mediated survival effects, SGK1 blocks apoptosis by preventing the de-attachment-induced dephosphorylation of Foxo3a (previously known as FKHRL1). 42 SGK1 suppresses Foxo3a transcriptional activity by phosphorylating its regulatory sites at Thr32 and Ser315, thus hinders Foxo3a-induced cell cycle arrest and apoptosis. 43 SGK1 is not only activated by AR but also regulates AR-mediated gene expression. SGK1 overexpression enhances intracellular AR (iAR) transactivation and promotes cell survival, even in the absence of androgen stimulation. 30 Membrane AR (mAR) has also demonstrated a strong iAR-independent tumor-inhibition effect. Inhibition of SGK1 enhances mAR-dependent apoptosis of breast cancer cells. 61 Moreover, the pro-survival and anti-apoptosis functions of SGK1 can also be implemented by inhibiting SEK1 binding to JUNK1 and MEKK1 via phosphorylation of SEK1 on Ser78, 40 upregulating oncogenic β-catenin,1,26 activating nuclear factor (NF)-κB transcriptional activity,49,62 promoting p53 degradation, 41 and increasing glucose uptake and ATP genesis. 63
SGK1 level is significantly higher in mesenchymal-subtype lung adenocarcinoma samples, based on RNA-seq data from The Cancer Genome Atlas (TCGA) database. 64 SGK1 inhibition attenuates epithelial-mesenchymal transition and metastasis of prostate cancer cells, while overexpression of SGK1 promotes their invasion and migration. 65 Similar results are observed in glioblastoma, colorectal, and hepatocellular carcinoma cells. Inhibition of SGK1 decreases the mesenchymal markers N-cadherin, vimentin, and focal adhesion kinase, and reduces the cell motility and invasion abilities. 18 As noted previously, mAR demonstrates strong antioxidant and antitumorigenic effects, which are mediated by vinculin phosphorylation and actin reorganization. Transfection with a constitutively active SGK1 mutant effectively dephosphorylates the cell-adhesion protein vinculin and enhances cell motility. 66 Interestingly, SGK1 has been shown to reduce cell migration and invasion. Lee et al. found that SGK1 favored colorectal cell differentiation and inhibited colorectal cancer metastasis in an orthotopic xenograft model. 28 Godbole et al. found that SGK1 expression was increased under progesterone treatment in breast cancer, which upregulated neuroblastoma-derived Myc downstream regulator gene 1 (NDRG1) via the AP-1 network. Increased NDRG1 expression reduced the activation of multiple cellular kinases and cell migration. 21 These inconsistent views may be due to the different models used, suggesting that SGK1 performs specific functions in different situations.
Autophagy
Autophagy is a crucial process in response to anti-tumor therapeutic stresses and is cytotoxic in certain circumstances. PI3K/mTOR and AMP-activated protein kinase are the central signaling pathways regulating autophagy, and SGK1 plays an intermediary role in that progress. 67 Inhibition of SGK1 by GSK650394 or shSGK1 promotes autophagy-dependent cell apoptosis via the mTOR-Foxo3a pathway, which is cytotoxic as the cell viability is restored by autophagy inhibitors. 31 Dual inhibition of mTOR and SGK1 enhances autophagy activation, thereby increasing the cytotoxic effect and inhibiting the progression of prostate cancer cells.31,65 A similar manifestation of cytotoxic autophagy is also observed in endometrial cancer cells 56 and glioblastoma multiforme cells after treatment with SGK1 inhibitors. 68
The conversion of LC3-I to LC3-II is considered as a marker of autophagy, 69 while GRP78, which is usually activated by endoplasmic reticulum stress, has an anti-apoptotic function and plays an important part in anti-radiation and chemotherapy. 68 SGK1 overexpression increases phosphorylation of endogenous Foxo3a at Thr32 and Ser257, and triggers the translocation of Foxo3a from the nucleus to the cytoplasm, preventing LC3-I/LC3-II conversion. 31 Treatment with SGK1 inhibitor leads to the conversion of LC3-I to LC3-II and decreases GRP78 and Bcl-2 expression, thereby releasing Beclin1 and inducing autophagy.31,56
Immunoregulation
The balance between Th1/Th2 cells is an important factor in anti-tumor immune responses. 70 SGK1 regulates the fate of T helper cells by acting as a positive regulator of Th2 differentiation and a negative regulator of Th1 differentiation. 46 SGK1 phosphorylates glycogen synthase kinase 3β (GSK-3β) in CD4+ helper T cells, thus blocking the degradation of β-catenin. 47 β-catenin, in turn, inhibits downstream targets associated with the Th1 phenotype by increasing the accumulation of T cell factor 1 (TCF1). 46 Moreover, SGK1 also phosphorylates Nedd4-2 at Ser221, Thr246, and Ser327, which inhibits the ubiquitination of JunB. The stability of JunB contributes to the expression of IL-4 and GATA-3, which is required by the Th2 cell lineage. 48 SGK1 deletion removes the inhibition of GSK-3β and Nedd4-2, thus promoting the Th1 and inhibiting the Th2 phenotype. 47
Th1 cells function as the main anti-tumor effector cells and are associated with prolonged survival in different types of tumors.71,72 Heikamp et al. generated T-Sgk1−/− mice in which SGK1 was selectively deleted in CD4+ helper precursor cells, and these mice demonstrated decreased IL-4 and inappropriate IFN-γ release. 46 Injection of B16 melanoma cells into wild-type and T-Sgk1−/− mice resulted in increased IFN-γ production and robust immunologic rejection of the tumors.46,73 This suggests that SGK1 inhibitors might have therapeutic value as an immunoadjuvant to tumor therapy by enhancing the Th1-mediated immune response.
In addition to the balance between Th1 and Th2 cells, SGK1 can also affect the balance between Th17 and Treg cells. SGK1 is a significant node of IL-23 signaling and promotes IL-23 receptor expression by phosphorylating Foxo1 and reducing its nuclear exclusion. 44 The pathogenic effector functions of Th17 cells are enhanced by the IL-23 receptor, and SGK1 is thus involved in IL-23-mediated enhancement of Th17 cell differentiation. 74 Foxp3 is also a critical regulator in Treg cell development and function, and its expression is also controlled by Foxo1 activity. 45
Th17 cells and Treg cells have been shown to play dual roles in cancer. Th17 cells accelerate tumor progression by promoting angiogenesis and immunosuppression, but also have an anti-tumor effect by recruiting immune cells, stimulating the effect of CD8+ T cells, and promoting transdifferentiation towards Th1 phenotypes. 73 Treg cells are generally thought to maintain immunological self-tolerance and suppress anti-tumor responses75–77; however, some reports also suggested that Treg cells might repress inflammation-driven tumorigenesis. 78 Besides, the Th17 switch mediated by SGK1 contributes to multiple chronic inflammatory diseases, including ulcerative colitis. Spagnuolo et al. found that the lymphocytes with high SGK1 level downregulated the SGK1 expression in colonic epithelial cells via the SGK1/Th17/Th13 axis and attenuated the cell viability. 79 This finding suggests a potential correlation between inflammatory disease and malignancy mediated by SGK1. More studies are therefore needed to clarify the effects of SGK1 in regulating the fate of immune cells, to improve its role as a potential target in cancer immunotherapy.
Ca2+ signaling
Ca2+ is an important intracellular messenger, and Ca2+ wave acts as a code for information transfer, contributing to the regulation of a range of cellular functions. 80 Mechanisms for increasing intracellular Ca2+ concentration include their release from intracellular stores and transport across the cell membrane.81,82 The depletion of Ca2+ in the endoplasmic reticulum activates the CRAC channel, resulting in store-operated Ca2+ entry. 82
ORAI and STIM are the key proteins that constitute CRAC channel: ORAI forms highly Ca2+-selective ion channels in the plasma membrane and is regulated by STIM, an endoplasmic reticulum-located Ca2+ sensor. 83 The most widely studied isoforms are ORAI1 and STIM1, which can be regulated by diverse kinases including SGK1.59,84 Sustained upregulation of store-operated Ca2+ entry by SGK1 through ORAI1 and STIM1 alters the Ca2+ code, which in turn impacts on tumor cell survival, proliferation, migration, and vascularization.85–88
SGK1 enhances ORAI1 by phosphorylating Nedd4-2. Nedd4-2 is an ubiquitin ligase mediating the degradation of many channel proteins including ORAI1, while phosphorylated Nedd4-2 binds to the protein 14-3-3, making it unable to ubiquitinate ORAI1. 89 SGK1 also stimulates the expression of ORAI1 and STIM1 by upregulating NF-κB.59,90 SGK1 phosphorylates IκB kinase β at Ser177 and Ser181, leading to the degradation of IκB and NF-κB nuclear translocation. 49 SGK1 can also affect NF-κB via phosphorylation of P300 at Ser1834 to enhance NF-κB acetylation. 50
Cancer stem cells
CSCs comprise a subgroup of tumor cells with the capacities for self-renewal and plasticity, analogous to stem cells. CSCs have shown an essential role in tumorigenesis, epithelial-mesenchymal transition, and chemoresistance.91,92 SGK1 acts as a major effector of mTORC2 in colorectal CSCs, and SGK1 knockdown significantly decreases the clonogenicity, invasion, and drug sensitivity of colorectal CSCs. 93 The CSC marker CD44 is decreased by inhibition of SGK1 in head and neck squamous cell carcinoma and prostate cancer.94,95 Using large-scale RNAi screens, Kulkarni et al. discovered that depletion of SGK1 reduced the survival and increased apoptosis of glioblastoma multiforme stem-like cells (GBM-SCs), as well as patient-derived GBM-SC lines, whereas SGK1 knockdown had little effect on serum glioma lines in vivo and in vitro. 20 Undifferentiated GBM-SCs were selectively sensitive while differentiated cells were tolerant of SGK1 inhibition. 20
Wnt/β-catenin, Notch, and Hedgehog are typical signaling pathways involved in CSCs. 92 However, although SGK1 has been shown to participate in regulating Notch and Hedgehog signaling, its role in CSCs remains unclear.96,97 A recent study indicated that SGK1 mediated Wnt/β-catenin signaling and facilitated increases in the CSCs population among radioresistant prostate cancer cells. 94
The development of the CSCs theory provides a reasonable explanation for tumorigenesis, metastasis, and recurrence. 92 However, the definition of CSCs remains ambiguous, and CSCs-targeted drugs have not demonstrated successful therapeutic effects in clinical trials. CSCs are known to exhibit extensive cell plasticity and to respond to multiple signals.92,98 Given that SGK1 is involved in cross-talk among different pathways, its role in the regulation of CSCs deserves further investigation.
Cell cycle disorder
The cell-cycle disorder is a hallmark of cancer progression. The cyclin-dependent kinase (CDK) inhibitor p27 oversees the inhibition of the cyclin E-CDK2 complex in the G0 and G1 phase and the activation of cyclin D-CDKs in early G1 phase. 99 Furthermore, the reduced level of p27 is shown to be an independent indicator of poor prognosis in patients with breast cancer. 100 SGK1 shuttles dynamically between the nucleus (in S and G2/M phases) and the cytoplasm (in G1 phase). 101 SGK1 also phosphorylates p27 at Thr157 and hinders nuclear p27 import, resulting in TGF-β resistance and cell cycle deregulation, while SGK1 silencing leads to G2/M arrest and induces caspase-dependent apoptosis. 102 Foxo3a interacts with p27 and downregulates its expression after phosphorylation by SGK1. 31
The tumor suppressor p53 is essential for preventing tumor formation by inducing cell cycle arrest and apoptosis. SGK1 phosphorylates MDM2 at Ser166 and mediates MDM2-dependent p53 ubiquitination and proteasomal degradation.41,103 Furthermore, p53 degradation occurs not only via MDM2 but also via Nedd4-2, which is reported to link with SGK1 through SEK1. 22 SGK1-induced p53 degradation reveals the effect of chronic psychological stress on cell proliferation, survival, and differentiation. 103 Interestingly, p53-dependent activation of ERK1/2 in response to DNA damage can also upregulate SGK1. 8
Chemo- and radio-resistance
Recent publications have reported that SGK1 can induce chemical and radiological resistance during the treatment of a variety of human tumors. 104 The PI3K-AKT-mTOR signaling pathway is known to be one of the most important pathways for cell survival, growth, proliferation, invasion, apoptosis inhibition, and angiogenesis, and inhibitors of this pathway have been widely investigated in preclinical studies and clinical trials. However, resistance to inhibitors targeting this pathway has recently emerged.23,39 SGK1 shares about 54% identity in the catalytic domain with Akt and is indicated as an important Akt-independent effector in PI3K/mTOR pathway. 3
SGK1 overexpression is associated with the resistance to Akt inhibitors and temozolomide (TZM) through phosphorylation of NDRG1. Sommer et al. found that Akt-inhibitor-resistant strains of breast cancer cells from two different clinical trials showed significantly elevated SGK1 levels, compared with Akt-inhibitor-sensitive cell lines. 23 They also observed a high degree of phosphorylation of NDRG1 in Akt-inhibitor-resistant cell lines. 23 NDRG1 reveals a pleiotropic function, as it is downregulated in colon, prostate, and breast cancers, and acts as a metastasis suppressor, 25 while it is upregulated in hepatic and gastric cancer and performs a pro-oncogenic function. 105 NDRG1 is a specific substrate of SGK1 and can be robustly phosphorylated by SGK1 at Thr328, Ser330, Thr346, Thr356, and Thr366. 106 Among these, phosphorylation at Ser330 and Thr346 determine the cellular distribution and activity of NDRG1. 35 Weiler et al. reported that SGK1-triggered phosphorylation of NDRG1 at Thr346 was increased in a glioma cell line resistant to TZM alkylating chemotherapy. 36 NDRG1 interacts with the DNA-repair enzyme O6-methylguanine-DNA methyltransferase, which is considered being associated with the resistance to alkane chemotherapies, promoting its stability and activity, while treatment with the SGK1 inhibitor EMD638683 overcomes NDRG 1-mediated TZM tolerance. 36
In addition, a high level of SGK1 induces resistance to PI3Kα inhibitors. SGK1 does not contain the PH domain, which is essential for Akt in PI3K-dependent membrane translocation recruited by phosphatidylinositol-(3,4,5)-triphosphate (PIP3), thus SGK1 is PIP3 independent and remains active even if PIP3 levels are negligible. 4 Castel et al. demonstrated that SGK1 could sustain Akt-independent mTORC1 activation under the circumstance of PI3Kα inhibition. 39
SGK1 can also induce paclitaxel resistance via modulation of RAN-binding protein 1 (RANBP1) expression. Paclitaxel administration results in an increase in prometaphase-blocked cells in SGK1-silenced RKO colon carcinoma cells, while this cell subset is decreased by SGK1 overexpression at the same paclitaxel concentration. Amato et al. found that Sp1 was phosphorylated by SGK1 at Ser59 and thus enhanced RANBP1 expression. 37 SGK1 inhibition significantly reduces nuclear transport by Sp1-dependent reduction of RANBP1 expression.37,38 Cells with RANBP1 interference results in an increased response to taxol-induced apoptosis, compared with those with normal or high RANBP1 levels. 107 A preclinical study of ovarian cancer xenografts model indicated that co-treatment of SGK1 inhibitor and paclitaxel significantly reduces the RANBP1 expression and restores paclitaxel sensitivity in paclitaxel-resistant murine. 108
The inhibition of SGK1 also increases the sensitivity to radiation treatment in vitro. For instance, the administration of an SGK1 inhibitor significantly increases the apoptosis after radiotherapy in colon carcinoma, 60 while the incubation of testosterone albumin conjugates with an SGK1 inhibitor enhances radiation-induced apoptosis in breast cancer. 61 SGK1 inhibition also significantly increases the effectiveness of radiation by augmenting phosphatidylserine exposure, caspase 3 activity, and DNA fragmentation but decreasing mitochondrial potential. 60 The combined use of SGK1 inhibitor and copper-64 shows an additive effect in tremendously increasing the level of human copper transporter 1 (CTR1), thus bringing a higher bioavailability of copper-64 in glioblastomas. 109 Co-treatment with SGK1 inhibitor reveals a potential reducing the cumulative toxicity of ionizing radiations, which represents a novel therapeutic alternative in tumors sensitive to radiation therapy. Although corresponding phenomena have been observed, the detailed mechanism by which SGK1 affects radiosensitivity remains to be clarified.
Clinical potential of SGK1 as a therapeutic target
Continuing research into the function of SGK1 has highlighted the clinical potential of this kinase. Under normal physiological conditions, SGK1 is expressed at low levels in several tissues, and a lack of SGK1 in SGK1−/− mice had no significant effect on survival. 110 However, SGK1 is upregulated under stress and certain pathological conditions and is thus regarded as a novel therapeutic target. 111
Akt, SGK1, and two other SGK isoforms, SGK2 and SGK3, show extensive overlap in terms of downstream targets and thus compensate for each other in gene ablation or depletion studies. 112 The discovery of selective SGK1 inhibitors has helped to identify SGK1-specific downstream targets and has provided invaluable tools for further clinical investigations (Table 2).
Summary of frequently or recently reported SGK1 inhibitors.

SGK1, serum and glucocorticoid-inducible kinase 1.
The heterocyclic indazole derivative later defined as GSK650394 is the first SGK1 inhibitor. 29 Berdel et al. demonstrated that GSK650394 decreased the expression of CD44 and HER2, thus suppressing tumor growth and tumorigenicity, while the combined application of GSK650394 and cisplatin was better than cisplatin alone in squamous cell carcinoma of the head and neck. 95 Wang et al. found that SGK1 played an antioxidant role through c-JUN-dependent NRF2 expression and activation in cervical cancer. The combination of GSK650394 and melatonin substantially repressed cervical tumors in vivo. 119 Besides, GSK650394 has been commercialized and its inhibitory effect is widely recognized, including tumor suppressor functions in pulmonary and prostate cancer.53,113 However, GSK650394 reveals notable off-target effects, that is it equivalently inhibits SGK2 as well and is only about 30-fold more selective for SGK1 than for other targets such as Akt, Rho-associated protein kinase, and Janus kinase isoforms (JAK1, JAK3). 104
The benzohydrazide derivative EMD638683 is another commonly used SGK1 inhibitor with a slightly higher selectivity. 114 Low-dose EMD638683 promotes radiation-induced apoptosis of colon tumor cells, 60 decreases the viability of rhabdomyosarcoma cells, and enhances the tumor-suppressive effect of doxorubicin. 115 However, EMD638683 shows poor cell permeability, and it may also inhibit cAMP-dependent protein kinase, mitogen- and stress-activated protein kinase 1, protein kinase C-related kinase, and other SGK isoforms. 114
Schenone et al. synthesized SI113 based on a pyrazolo[3,4-d]pyrimidine scaffold. 116 SI113 displays high selectivity for inhibiting SGK1 kinase activity by competing with ATP for the binding domain and is much less effective against Akt1. 116 Thus, the short-term systemic toxicity of SI113 is low in preclinical observation. In neurospheres, SI113 induces pro-survival autophagy by reducing the phosphorylation of SGK1 and mTOR activity and increasing the AMPK activity. SI113 and quinacrine, an autophagy inhibitor, show a strong synergistic effect in hindering the tumor growth. 120 In colon cancer, hepatocellular carcinoma, and glioblastoma multiforme, SI113 is capable of inhibiting cancer cell malignancy by blocking focal adhesion kinase (FAK), EMT, and cytoskeletal organization. 18 The tumor-killing effects of radiotherapy and paclitaxel are potentiated by synergy with SI113.121,122
In addition to the three mentioned previously, new inhibitors, including SGK1-inh and QGY-5-114-A, are ceaselessly being designed and synthesized.117,118 However, their effectiveness in tumor cells remains to be further investigated.
Increasing evidence suggests that SGK1 inhibitors represent a novel tumor therapy approach, extending traditional strategies. On that basis, the structure and function of inhibitors need to be improved, and in vivo pharmacodynamic and pharmacokinetic data are required to conduct clinical trials.
Conclusion
SGK1 is a pleiotropic factor involved in several physiological and pathological processes and has shown important effects on oncology. As an essential Akt-independent mediator of the PI3K/mTOR signaling pathway, SGK1 is transcriptionally and posttranslationally affected by various factors, including hormones, cytokines, and stress response. SGK1 is involved in regulating tumor growth, survival, metastasis, autophagy, immunoregulation, Ca2+ signaling, cancer stem cells, cell cycle, and mediates the therapeutic resistance. SGK1 inhibition is regarded as a potential tumor therapeutic approach, but further pharmacological investigations are needed to advance clinical trials of SGK1-targeted therapies.
Footnotes
Author contributions
All authors contributed to the writing and editing of this manuscript. All authors read and approved the final manuscript.
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by grants from the National Natural Science Foundation of China (No. 81772639; 81802475; 81972258; 81974376); Natural Science Foundation of Beijing (No. 7192157); CAMS Innovation Fund for Medical Sciences (CIFMS) (No.2016-I2M-1-001); China Postdoctoral Science Foundation (No.198831); National Key R&D Program of China (2018YFE0118600); Non-profit Central Research Institute Fund of Chinese Academy of Medical Sciences (2019XK320001).