
Abstract
Epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs) are the currently recommended treatment for advanced EGFR mutation-positive non-small cell lung cancer (NSCLC). Acquired resistance inevitably develops, with the EGFR T790M mutation comprising approximately 55% of the mechanisms of resistance following first- or second-generation EGFR-TKI therapy (e.g. gefitinib, erlotinib, afatinib, and dacomitinib). Patients without T790M are a heterogeneous group for whom platinum-based chemotherapy is currently recommended as a second-line treatment. In addition to secondary mutations in EGFR (e.g. T790M), the currently known resistance mechanisms can be classified into the following three categories: bypass pathways, downstream signaling pathways, and histologic transformations. Given the evolving knowledge and convenience of diagnosing acquired resistance mechanisms by next-generation sequencing and liquid biopsy, exploratory studies targeting these resistance mechanisms and incorporating immunotherapy into the treatment paradigm have become the mainstream of future development. This review focuses on acquired resistance mechanisms other than T790M that develop after first- or second-generation EGFR-TKI therapy. Exploratory second-line treatments targeting resistance mechanisms as well as combination immunotherapy and chemotherapy in ongoing clinical trials are reviewed here. We also highlight the recent development of next-generation sequencing and liquid biopsy in this field.
Introduction
First- and second-generation epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs), including gefitinib, erlotinib, afatinib, and dacomitinib, are effective as first-line treatment for advanced non-small cell lung cancer (NSCLC) harboring activating EGFR mutations (e.g. deletions in exon 19 and the exon 21 L858R mutation).1–7 EGFR T790M mutation emerges following EGFR-TKI therapy, and accounts for 55% of mechanisms of acquired resistance to first- and second-generation EGFR-TKIs.8–11 Osimertinib monotherapy is the currently recommended second-line treatment for EGFR T790M mutation-positive (T790M-pos) NSCLC.12–14 Other secondary resistance mutations in EGFR, such as D761Y, T854A, and L747S, are rare, and the irreversible EGFR-TKIs, afatinib and osimertinib, have been shown to inhibit EGFR phosphorylation in cells harboring these secondary mutations.15–18 For patients with EGFR T790M mutation-negative (T790M-neg) NSCLC, platinum-based chemotherapy is the currently recommended second-line treatment.19–21 In addition to the T790M resistance mutation, the molecular alternations identified as resistance mechanisms include bypass pathway activation [e.g. MET amplification [MET-amp] and HER2 amplification (HER2-amp)] and downstream signaling pathways (e.g. PI3K and BRAF mutations). Histological transformations [e.g. small cell and epithelial–mesenchymal transition (EMT)] are also mechanisms of resistance. T790M-neg NSCLC comprises these mechanisms plus other unknown mechanisms and is seen in a heterogeneous group of patients. In the era of molecular targeted therapy, immunotherapy, next-generation sequencing (NGS), and liquid biopsy, exploratory strategies are under development to identify patients suitable for molecular targeted therapy to overcome resistance mechanisms.
This review describes recent developments in the second-line treatment of advanced T790M-neg NSCLC following first- and second-generation EGFR-TKI therapy. We assess the role of molecular-targeted agent combinations, immunotherapy-chemotherapy combinations, and other treatment strategies, with a focus on those discussed in prospective clinical trials. None of these exploratory treatments has received approval for advanced T790M-neg NSCLC. A literature review of clinical studies published between July 2017 and June 2019 was conducted in PubMed and MEDLINE using the keywords ‘non-small cell lung cancer,’ ‘T790M-negative,’ ‘EGFR mutation,’ ‘acquired resistance,’ and ‘immune checkpoint inhibitor.’ We also performed a manual search of abstracts from presentations at major oncology meetings.
Mechanisms of acquired resistance and exploratory treatments: bypass pathways
MET amplification
MET-amp is present in 5–20% of EGFR mutation-positive NSCLC patients who develop acquired resistance to EGFR-TKIs (Figure 1).8,22–25 Patients who harbor preexisting MET-amp before EGFR-TKI therapy may have a shorter time to progression on EGFR-TKI therapy.26,27 MET-amp reactivates the PI3K/AKT pathway mediated by ErbB3 transactivation; inhibition of MET signaling could restore the sensitivity of lung cancer cells to gefitinib. 22 Overexpression of hepatocyte growth factor (HGF), the ligand of MET, is also implicated in inducing resistance to EGFR-TKIs, and high serum levels of HGF is a poor prognostic factor in NSCLC patients treated with EGFR-TKIs.28–30 Both MET-amp and HGF overexpression can occur with or without T790M.22,23 Based on these findings, MET inhibitors were evaluated to overcome this resistance mechanism.

Estimated distribution of resistance mechanisms to first- and second-generation EGFR-TKIs and related exploratory treatments (red boxes).
Multi-kinase inhibitors
Crizotinib is an anaplastic lymphoma kinase (ALK), MET, and ROS1 inhibitor. Crizotinib monotherapy and combination therapy with an EGFR-TKI were reported preclinically and clinically to overcome the MET-amp-mediated primary or acquired resistance to EGFR-TKI therapy.31–33 A phase I study of crizotinib plus erlotinib therapy reported maximum tolerated doses of crizotinib and erlotinib of 150 mg twice daily and 100 mg daily, respectively. However, there is no ongoing clinical trial of crizotinib in combination with EGFR-TKI for treatment of this MET-amp patient population. 34
A phase II study evaluated erlotinib (150 mg daily) plus cabozantinib [a MET, vascular endothelial growth factor 2 (VEGFR2), RET, AXL, and KIT inhibitor] (40 mg daily) in advanced EGFR mutation-positive NSCLC patients pretreated with EGFR-TKIs. Most of the patients had received afatinib therapy and experienced disease progression. The objective response rates (ORRs) were 10.8% among the 37 subjects, 0% in 7 tissue T790M-neg subjects, and 17.6% in 17 plasma T790M-neg subjects. None of the patients in this cohort harbored MET-amp. Thus, the group of patients benefitting from this treatment remains unknown. 35 A phase I/II study of S 49076 [a MET/AXL/fibroblast growth factor receptor (FGFR) inhibitor] is ongoing, which combines gefitinib for patients with first- and second-generation EGFR-TKI pretreated EGFR mutation-positive/T790M-neg NSCLC with MET or AXL dysregulation (Clinicaltrialsregister.eu, EudraCT number: 2015-002646-31). 36
Selective MET inhibitors
Tivantinib (ARQ 197) is a selective MET inhibitor. A phase II study conducted in Japan enrolled patients with advanced EGFR mutation-positive NSCLC who developed acquired resistance to gefitinib or erlotinib to receive tivantinib plus erlotinib therapy. A total of 45 patients were enrolled, half of whom were T790M-pos, with an ORR of 6.7%. High MET expression (⩾50%) by immunohistochemical (IHC) staining was detected in 48.9% of the patients, including all three responders (Table 1). 37
Capmatinib is another selective MET inhibitor. Preclinical studies of capmatinib in combination with other kinase inhibitors demonstrated its activity to overcome MET-driven resistance.41,42 A phase Ib/II study enrolled EGFR-TKI pretreated, T790M-neg patients to receive gefitinib and capmatinib combination therapy. In phase II, 100 patients received gefitinib (250 mg daily) and capmatinib (400 mg twice per day), with ORRs in patients with MET-amp [gene copy number (GCN) ⩾ 6, n = 36] and MET overexpression (IHC 3+, n = 78) of 47% and 32%, respectively (Table 1). The authors concluded that MET detection by fluorescent in situ hybridization (FISH) using a cutoff value of GCN ⩾ 6 may identify the group of patients who may benefit from this treatment. 38 An ongoing phase Ib/II GEOMETRY duo-1 study (ClinicalTrials.gov identifier: NCT02468661) aims to compare capmatinib monotherapy, capmatinib plus erlotinib, and platinum plus pemetrexed chemotherapy in EGFR-TKI pretreated EGFR mutation-positive NSCLC patients positive for MET-amp. 43 Another phase Ib/II study combines nazartinib (EGF816, a third-generation EGFR-TKI) and capmatinib in EGFR mutation-positive NSCLC patients (ClinicalTrials.gov identifier: NCT02335944) in various treatment settings and T790M/MET status.
Selected clinical efficacy reports of selective MET inhibitors.

AE, adverse event; CI, confidence interval; EGFR, epidermal growth factor receptor; GCN, gene copy number; NA, not available; NSCLC, non-small cell lung cancer; ORR, objective response rate; PFS, progression-free survival; TKI, tyrosine kinase inhibitor.
For each AE, the reported values in this column are the percentages of patients receiving the therapy who experienced the AE, and the percentage of patients receiving the therapy who experienced the AE at grade ⩾3).
A Japanese phase I study recommended tivantinib (240 mg twice daily) in patients homozygous for CYP2C19 loss-of-function polymorphisms (poor metabolizers), and 360 mg twice per day in the other patients (extensive metabolizers).
Tepotinib is another selective MET inhibitor. A phase II study randomized EGFR-TKI pretreated, T790M-neg, MET IHC2+/3+ and MET-amp patients to receive gefitinib (250 mg daily) and tepotinib (500 mg daily) combination therapy or platinum-pemetrexed chemotherapy. The ORRs in patients with MET IHC 3+ (n = 19) and MET GCN ⩾ 5 (n = 12) were 68.4% and 66.7%, respectively (Table 1). 39
Another selective MET inhibitor, savolitinib, was tested in the TATTON study (ClinicalTrials.gov identifier: NCT02143466) in combination with osimertinib in patients with pretreated EGFR mutation-positive NSCLC. In this trial, osimertinib (80 mg daily) and savolitinib (600 mg daily) were administered in a cohort of first- and second-generation EGFR-TKI pretreated patients with T790M-neg/MET-positive (MET GCN ⩾ 5 or IHC 3+) disease. The ORR was 52% (n = 46) and the median duration of response was 7.1 months (Table 1). 40 The ORR was 25% (n = 48) and the median duration of response was 9.7 months in another cohort within this study that included third-generation EGFR-TKI pretreated patients administered the combination therapy. Future development of this combination therapy is aimed at osimertinib-pretreated, MET-positive, and EGFR mutation-positive NSCLC patients (ClinicalTrials.gov identifier: NCT03778229).
Anti-MET antibodies
Emibetuzumab (LY2875358) is a humanized IgG4 monoclonal bivalent MET antibody. A study that enrolled patients with advanced NSCLC (enriched for EGFR mutation-positive patients) who developed acquired resistance to erlotinib to receive emibetuzumab with or without erlotinib therapy reported ORRs among patients with MET overexpression (⩾60%) of 3.8% and 4.8% in the combination and monotherapy arms, respectively (ClinicalTrials.gov identifier: NCT01900652). 44 The authors concluded that these therapies did not reverse resistance.
Telisotuzumab vedotin (ABBV-399; teliso-v) is an anti–c-MET antibody and monomethyl auristatin E drug conjugate. A phase Ib study combining ABBV-399 with erlotinib for MET-positive patients defined as centrally confirmed IHC H– score >150 or locally confirmed MET-amp (ClinicalTrials.gov identifier: NCT02099058) reported an ORR of 34.5% in 29 EGFR-TKI pretreated (first-, second-, or third-generation) patients. Grade ⩾3 pulmonary embolism developed in 14% of the subjects. 45
Despite the active development of MET inhibitors in this post-EGFR-TKI therapy setting, none of the MET inhibitors or drugs described above is approved for patients with EGFR-TKI pretreated EGFR mutation-positive, MET-amp NSCLC. In the aforementioned studies, enrolling T790M-pos patients (e.g. the tivantinib trial), various definitions of MET activation (e.g. the capmatinib trial) and slow patient accrual (e.g. the tepotinib trial) potentially made these studies inconclusive.
Human epidermal growth factor receptor 2 (HER2) amplification
Restoration of EGFR downstream signaling pathways by HER2 amplification (HER2-amp) occurs in about 10% of cases with acquired resistance, and is mutually exclusive with the T790M mutation (Figure 1).8,46 HER2, like EGFR, belongs to the ErbB family of receptor tyrosine kinases and can form heterodimers with EGFR to activate downstream signaling. Patients with preexisting HER2-amp before EGFR-TKI therapy may have a shorter time-to-progression on EGFR-TKI therapy. 26 In a genetically modified mouse model, osimertinib showed activity against EGFR-deletion 19/HER2 coexpression lung cancer. 47 A phase II study evaluating the efficacy of trastuzumab emtansine (T-DM1) in patients with HER2 overexpressing (tested with IHC stain, 2+ or 3+) metastatic NSCLC enrolled 49 patients, 20 of whom had HER2 IHC3+, including all four responders (ORR: 20% among IHC 3+ patients). 48 One responder had EGFR mutation-positive disease pretreated with erlotinib and HER2-amp (archival tissue). The progression-free survival (PFS) of this patient was 9.6 months. The authors concluded that HER2 IHC alone was an insufficient predictive biomarker for T-DM1 activity, and that additional investigation of HER2 overexpression, amplification, and mutation may help to identify patient populations that might benefit from this treatment. 48 None of the HER2 targeting agents is approved for patients with EGFR-TKI pretreated, EGFR mutation-positive, HER2-amp NSCLC.
AXL
Activation of the receptor tyrosine kinase AXL and its ligand GAS6 is another mechanism of acquired resistance to EGFR-TKI therapy, especially erlotinib, in EGFR mutation-positive NSCLC. Inhibition of AXL restored sensitivity to erlotinib therapy. 49 AXL and EGFR share the same PI3K/AKT and MAPK/ERK downstream signaling pathway and AXL-mediated resistance involved in the activation of both pathways. 50 AXL expression increased EMT, which was also a mechanism of EGFR-TKI resistance. An AXL inhibitor and erlotinib combination treatment reversed erlotinib resistance in mesenchymal cell lines expressing AXL.51,52 Clinical trials of AXL inhibitor monotherapy or in combination with an EGFR-TKI to overcome resistance are ongoing, including TP-0903 monotherapy (ClinicalTrials.gov identifier: NCT02729298), BGB324 plus erlotinib (ClinicalTrials.gov identifier: NCT02424617), DS-1205c plus osimertinib (ClinicalTrials.gov identifier: NCT03255083), and DS-1205c plus gefitinib (ClinicalTrials.gov identifier: NCT03599518). The two DS-1205c studies are aimed at T790M-neg patients.
Other bypass pathways
Preclinical and clinical studies have explored other bypass pathways, such as Interleukin-6/Janus kinase/signal transducers and activators of transcription (IL-6/JAK/STAT) signaling,53–56 insulin-like growth factor-1 receptor (IGF1R) pathway,57–59 and FGFR3-TACC3 fusion. 60 A phase Ib/II study evaluated erlotinib plus ruxolitinib (a JAK 1/2 inhibitor) in patients with erlotinib-pretreated EGFR mutation-positive NSCLC. Among 22 enrolled patients, 18 of whom were T790M-neg, there was only one responder, who was T790M-pos. 61 Another phase Ib study conducted in Korea evaluating afatinib plus ruxolitinib for EGFR-TKI pretreated, EGFR mutation-positive NSCLC patients reported an ORR of 20% in 10 T790M-neg subjects. 62
Mechanisms of acquired resistance and exploratory treatments: downstream signaling
RAS/RAF/MEK/ERK pathway
BRAF mutations (e.g. V600E and G469A) are the mechanism of acquired resistance to EGFR-TKI therapy in 1% of patients with EGFR mutation-positive NSCLC (Figure 1). 63 Dabrafenib (a BRAF inhibitor) and trametinib (a MEK inhibitor) combination therapy has been approved for treatment in patients with metastatic BRAF V600E mutation-positive NSCLC. 64 However, the efficacy of this combination therapy in EGFR-TKI pretreated EGFR mutation-positive patients is not known. A recent study demonstrated that rare BRAF kinase fusions are a potentially targetable mechanism of resistance to EGFR-TKIs. 65 Other strategies to overcome resistance in this pathway included dual inhibition of MEK and EGFR, which acts by inhibiting ERK phosphorylation and upregulating the expression of the proapoptotic protein BIM.66,67 Selumetinib (a MEK inhibitor) is undergoing development in combination with osimertinib in many different clinical settings. In the TATTON study (ClinicalTrials.gov identifier: NCT02143466), a small cohort of 12 EGFR mutation-positive NSCLC patients pretreated with first- and second-generation EGFR-TKIs were administered osimertinib plus selumetinib therapy. The ORR was 67% (n = 12) and all three T790M-neg patients achieved a partial response. 68 A prospective clinical trial of selumetinib and gefitinib combination therapy for advanced EGFR-TKI pretreated EGFR mutation-positive NSCLC has completed patient accrual (ClinicalTrials.gov identifier: NCT02025114). Future development of this combination aims at first-line treatment in advanced EGFR mutation-positive NSCLC patients (ClinicalTrials.gov identifier: NCT03392246).
PI3K/AKT/mTOR pathway
PIK3CA mutations frequently coexist with EGFR mutations and are a mechanism of acquired resistance to EGFR-TKI therapy in 0–5% of EGFR mutation-positive NSCLC patients (Figure 1).24,69 Targeting of the PI3K/AKT/mTOR pathway in NSCLC has long been under development. 70 Early development of buparlisib monotherapy (BKM120, a pan-PI3K inhibitor) for PI3K pathway-activated NSCLC patients (PI3K mutation, PTEN mutation, or PTEN negative by IHC) showed a poor 12-week PFS rate, suggesting that the combination of buparlisib with other agents may demonstrate better efficacy. 71 However, formal reports of further development of buparlisib in combination with EGFR-TKIs (ClinicalTrials.gov identifiers: NCT01487265 and NCT01570296) are not yet available. 72 MK-2206 is an AKT inhibitor that, in combination with erlotinib, showed an ORR of 9% in 45 EGFR mutation-positive NSCLC patients pretreated with EGFR-TKIs. 73 A preclinical study showed that collagen type I induced EGFR-TKI resistance via mTOR activation, independent of AKT. 74 MTOR mutations were identified recently as a mechanism of acquired resistance. 26 An ongoing study is assessing sapanisertib (TAK-228, an mTORC1/2 inhibitor) in combination with osimertinib for EGFR mutation-positive/T790M-neg NSCLC patients pretreated with first- and second-generation EGFR-TKIs (ClinicalTrials.gov identifier: NCT02503722).
Mechanisms of acquired resistance and exploratory treatments: histologic transformation
Small cell transformation
The transformation from EGFR-mutated adenocarcinoma to small cell lung cancer leads to acquired resistance in 5–15% of patients, who also show aggressive clinical progression (Figure 1).8,24 Retinoblastoma (RB) is lost in almost all cases of small cell transformation. 75 EGFR-TKI-resistant lung adenocarcinoma and small cell carcinoma share a common clonal origin and undergo branched evolutionary trajectories. EGFR mutation-positive lung adenocarcinoma with completely inactivated Rb and p53 had a greater risk of small cell transformation. 76 The median time to transformation was 17.8 months. Platinum-etoposide and taxanes yielded high response rates and a median OS of 10.9 months after small cell transformation.77,78 A study of patient-derived cell lines observed EMT phenotypes in small cell-transformed cells. Thus, adding AKT or histone deacetylase inhibitors to EGFR-TKI therapy may be effective. 79
Epithelial–mesenchymal transition
EMT in tumor cells promotes tumor invasion, metastasis, stem cell properties, and immunosuppression.80,81 The cancer cells lose epithelial features, such as E-cadherin expression, and transform to a spindle-like mesenchymal morphology with high vimentin/fibronectin expression. EMT is also a mechanism of acquired resistance to EGFR-TKIs in EGFR mutation-positive NSCLC. 82 The activation of several pathways and molecules involving EMT, including Slug; Snail; AXL; transforming growth factor (TGF)-β; interleukin (IL)-6; Notch-1; platelet-derived growth factor receptor (PDGFR); zinc finger E-box-binding homeobox 1 (ZEB1); mediator of RNA polymerase II transcription, subunit 12 homolog (MED12); integrin β3; LIN28B; BIM; Hedgehog pathway; and TWIST1 is associated with acquired resistance to EGFR-TKI.50,54,82–92 AXL inhibition is one treatment targeting EMT (see the AXL section). A recent preclinical study demonstrated that inhibition of TWIST1 or BCL2 may overcome EMT-mediated resistance to EGFR-TKIs. 90 Another preclinical study of EMT features-exhibiting gefitinib-resistant cells demonstrated that histone deacetylases and 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG-CoA) reductase inhibitor inhibited Src/Hakai and Hakai/E-cadherin interactions, reversed E-cadherin expression, and attenuated vimentin and stemness to restore gefitinib sensitivity. 93 A detailed review of targeted EMT signaling in lung cancer is available elsewhere. 94
Mechanisms of acquired resistance and exploratory treatments: others
Because EGFR is a client of chaperone heat shock protein 90 (HSP90), HSP90 inhibitors have been evaluated in efforts to overcome resistance to EGFR-TKI therapy. A phase I/II study administered AUY922 and erlotinib to EGFR mutation-positive NSCLC patients pretreated with EGFR-TKIs, reporting an ORR of 16% (n = 25); however, the duration of treatment was limited by adverse events (AEs), especially night blindness. There were three responders among nine T790M-pos patients, but only one responder among 15 T790M-neg subjects. 95 YES1 encodes the Src family kinase, and YES1 amplification was recently identified as a mechanism of acquired resistance to EGFR-TKIs.26,96 Kelch-like ECH-associated protein 1 (KEAP1) loss was also recently identified as a mechanism of acquired resistance to EGFR-TKIs. 26 A preclinical study using CRISPR-Cas9 gene deletion reported that the loss of KEAP1 increased NRF2 expression and modulated the response to many TKIs. Loss of KEAP1 altered cell metabolism and allowed cell proliferation in the absence of MAPK signaling. 97
Other exploratory second-line treatments
EGFR-TKI and anti-EGFR antibody combinations: vertical blockades
A phase Ib study including patients with advanced EGFR mutation-positive NSCLC pretreated with gefitinib or erlotinib were administered afatinib (40 mg daily) plus cetuximab (500 mg/m2, every 2 weeks). The ORR in the T790M-neg subgroup (n = 53) was 25%, while the duration of response and median PFS were 9.5 and 4.6 months, respectively. Overall, 13% of the patients discontinued treatment due to AEs, while 64% of the patients did not require a dose reduction. 98
A study of necitumumab (a human IgG1 monoclonal antibody against EGFR) plus osimertinib is ongoing for patients with EGFR mutation-positive NSCLC pretreated with first- and second-generation EGFR-TKIs (ClinicalTrials.gov identifier: NCT02496663).
EGFR-TKI and chemotherapy combination
The IMPRESS study was a phase III study comparing gefitinib or placebo plus cisplatin-pemetrexed chemotherapy after progression on gefitinib therapy in patients with advanced EGFR mutation-positive NSCLC. Overall, the PFS (the primary endpoint) was 5.4 months in both arms, with an inferior OS in the gefitinib plus chemotherapy arm (13.4 versus 19.5 months). Among patients with T790M-neg disease detected in plasma samples by bead, emulsion, amplification, and magnetics (BEAMing) digital polymerase chain reaction (PCR), the PFS was longer in those administered gefitinib plus chemotherapy (6.7 versus 5.4 months) while the OS was similar (21.4 versus 22.5 months). The authors concluded that the detrimental effect of continuing gefitinib in combination with chemotherapy may be driven by T790M-pos status; therefore, this approach is no longer recommended, regardless of T790M status.99,100
EGFR-TKI-immunotherapy combination
Activation of the programmed cell death protein 1/programmed cell death ligand 1 (PD-1/PD-L1) pathway mediated immune escape in EGFR mutation-positive NSCLC, while EGFR-TKI therapy downregulated PD-L1.101–103 In patients who developed resistance to EGFR-TKI therapy, T790M-neg tumors were associated with higher PD-L1 expression compared with that in T790M-pos tumors. 104 Another study reported increased PD-L1 expression in some patients who developed resistance to gefitinib therapy; moreover, MET IHC positivity was significantly associated with this group. 105 PD-L1 expression might confer resistance to EGFR-TKIs via the upregulation of YAP1 expression. 106 A retrospective study demonstrated that nivolumab monotherapy was more effective in treating T790M-neg compared with that in T790M-pos patients. 107 A phase I study enrolled patients with EGFR mutation-positive NSCLC pretreated with EGFR-TKIs who were administered nivolumab (3 mg/kg every 2 weeks) and erlotinib (150 mg daily) therapy. The ORR was 15% among the 20 subjects. Two of the eight T790M-neg subjects achieved a partial response, one of whom had MET-amp. 108 Despite recent developments in immune checkpoint inhibitors and EGFR-TKI combination therapy, no ongoing clinical trials have focused on T790M-neg patients.109,110
Immunotherapy-chemotherapy combination
Immune checkpoint inhibitors in combination with platinum-based chemotherapy are the currently recommended first-line treatment for advanced EGFR mutation-negative/ALK-negative NSCLC.111,112 In the IMpower150 study, atezolizumab plus bevacizumab plus carboplatin plus paclitaxel (ABCP) treatment significantly prolonged the median PFS (8.3 versus 6.8 months) and OS (19.2 versus 14.7 months) in EGFR mutation-negative/ALK-negative patients as compared with those in patients administered bevacizumab plus carboplatin plus paclitaxel (BCP) treatment. 111 Patients with EGFR mutations or ALK-positive NSCLC were originally allowed to participate in this study following the progression of EGFR-TKI and ALK inhibitor therapies, respectively. However, these patients were excluded from the primary analysis. In the subgroup of EGFR mutation-positive patients (n = 124), the median OS was not estimable in the ABCP group as compared with 18.7 months in the BCP group [hazard ratio (HR) 0.61, 95% confidence interval (CI) 0.29–1.28]. In patients harboring activating EGFR mutations (deletions in exon 19 or with the exon 21 L858R mutation), the median OS was not estimable in the ABCP group (n = 26) as compared with 17.5 months in the BCP group (n = 32) (HR 0.31, 95% CI 0.11–0.83). The authors concluded that the OS in patients with sensitizing EGFR mutations warranted further study. 113 However, the T790M status was not widely available in the subjects and the patient number was too low to make conclusions. ABCP has not yet received approval from the US Food and Drug Administration (FDA) for the treatment of EGFR mutation-positive NSCLC.
Ongoing studies are evaluating immunotherapy-chemotherapy combinations in T790M-neg NSCLC patients following prior first- and second-generation EGFR-TKI therapy (Table 2). However, the identification of a potentially predictive biomarker of treatment efficacy (e.g. PD-L1 expression and tumor mutation burden) will be challenging in the heterogeneous population of T790M-neg patients.
Ongoing clinical studies of combination immunotherapy and chemotherapy in pretreated EGFR-mutant NSCLC.
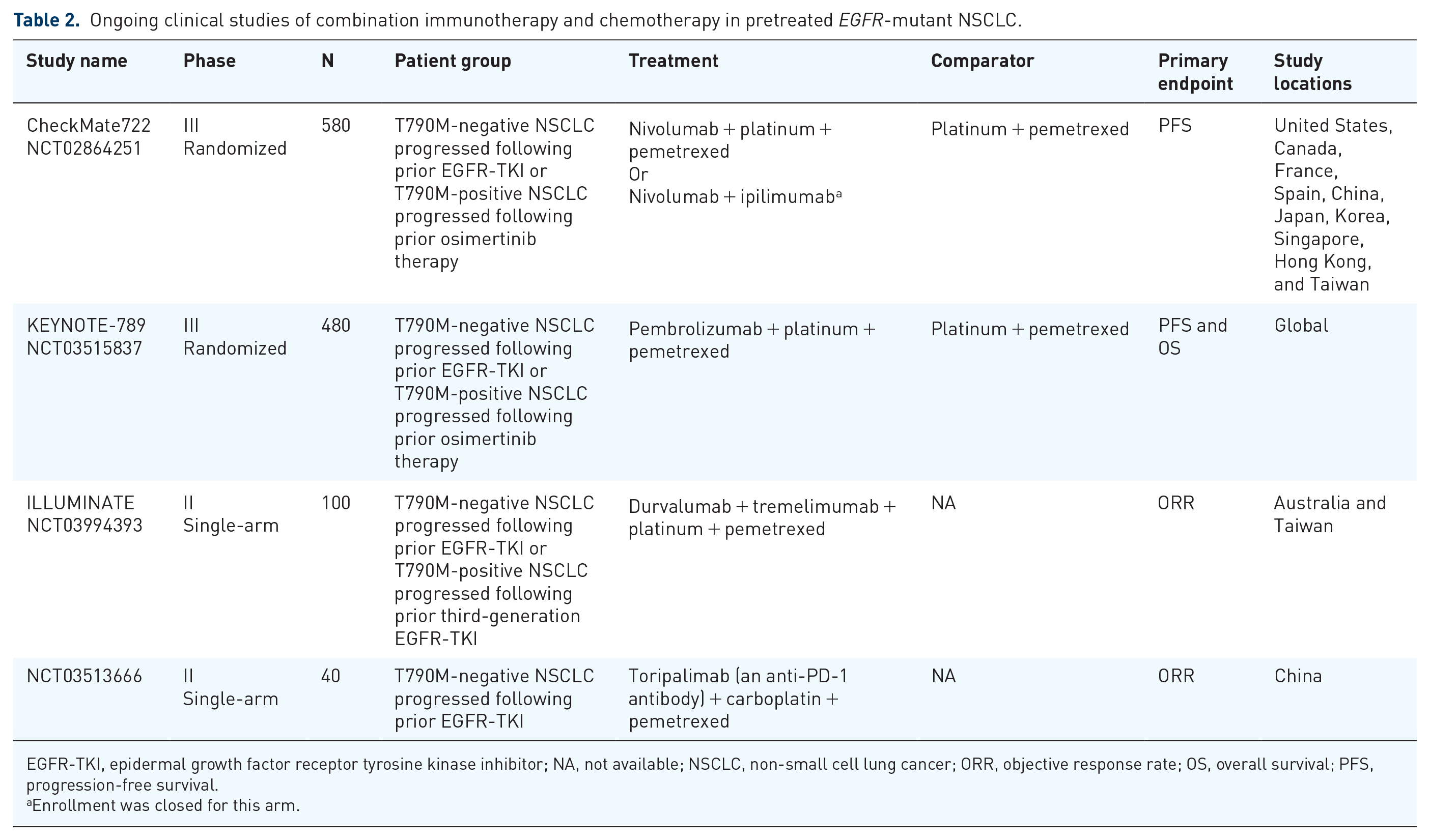
EGFR-TKI, epidermal growth factor receptor tyrosine kinase inhibitor; NA, not available; NSCLC, non-small cell lung cancer; ORR, objective response rate; OS, overall survival; PFS, progression-free survival.
Enrollment was closed for this arm.
Anti-HER3 antibody-drug conjugate
Heregulin, a ligand of HER3, activates HER3 in an autocrine fashion and causes erlotinib resistance. Patritumab (U3-1287, an anti-HER3 monoclonal antibody) and erlotinib combination therapy overcame resistance in a heregulin-overexpressed preclinical model. 114 U3-1402 is an antibody–drug conjugate that targets HER3 expression. This drug comprises an anti-HER3 antibody attached to a topoisomerase I inhibitor. A preclinical study showed that treatment with U3-1402 in gefitinib-resistant HCC827 cells with MET-amp induced apoptosis and phosphorylation of histone H2A.X (a marker of DNA damage) but did not inhibit HER3/PI3K/AKT signaling. These resistance cells had higher HER3 expression, which indicated increased internalization of U3-1402. 115 In a phase I study, enrolled patients with EGFR mutation-positive/T790M-neg NSCLC were pretreated with first- and second-generation EGFR-TKIs or osimertinib-pretreated patients (ClinicalTrials.gov identifier: NCT03260491). Among 23 (21 of whom had received osimertinib therapy) patients administered treatment, 4 of the 16 evaluable patients achieved a partial response. The median H-score of membrane HER3 expression was 193 among the 19 evaluable samples. 116
Diagnosis of the mechanisms of acquired resistance
Tissue-based diagnosis
The mechanisms of acquired resistance in T790M-neg NSCLC are heterogeneous; however, identification of the exact mechanism is crucial to guide second-line treatment. Rebiopsy of the resistant tumor to detect the resistance mechanism using the cobas® EGFR Mutation Test v2 (Roche Molecular Systems, Inc., Pleasanton, CA, USA) is the standard-of-care for EGFR-TKI pretreated EGFR mutation-positive NSCLC, at least for the diagnosis of T790M-pos disease. The yield rate of tumor rebiopsy in EGFR mutation-positive NSCLC depended on the tumor size, tumor accessibility, operator skill and experience, patient preference, and spatiotemporal heterogeneity.117–119 This method has the limitation that it detects only known resistance mechanisms related to the T790M mutation. NGS, especially the hybrid-capture based platform, is a new way to detect multiple genetic mutations and genomic signatures. The FoundationOne CDx® (F1CDx, Foundation Medicine, Inc.) is an FDA-approved tissue-based NGS-based in vitro diagnostic tool for solid tumors that has been used to discover some resistance mechanisms (e.g. BRAF, FGFR3-TACC3, and CCDC6-RET fusion).60,65,120 The Memorial Sloan Kettering–Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) assay is another hybridization capture, tissue-based NGS platform widely used for the study of lung cancer, which was used to discover YES1 amplification and MTOR mutations.26,121 A recent case report also discovered MET exon 14 skipping alteration as a mechanism of acquired resistance to EGFR-TKIs using this platform. 122 These diagnostic tools should be evaluated in prospective clinical trials to guide second-line treatment in patients with T790M-neg NSCLC. The analysis of NGS data by computational methods and machine learning techniques may predict polytherapy switching strategies to overcome tumor heterogeneity and evolution. 123
Liquid biopsy: plasma-based diagnosis
The sensitivity of plasma genotyping to detect T790M mutation was 77%, 70%, and 61% by droplet digital PCR, BEAMing, and cobas® plasma tests, respectively.124–127 Patients with negative plasma T790M findings should undergo tumor biopsy to determine the tissue T790M status for subsequent therapy. 124 In patients with T790M-neg NSCLC, which is a heterogeneous group, analysis of plasma circulating tumor DNA (ctDNA) may reveal a comprehensive genetic landscape of the tumors that developed acquired resistance. Guardant360 (Guardant Health) is a plasma-based NGS test that has shown a high concordance with tissue genotyping in untreated NSCLC patients. 128 It is also useful for detecting resistance mechanisms and possibly guiding subsequent therapy.129,130 As mutational subclones may evolve over time, liquid biopsy allows for real-time monitoring of clonal changes. Prospective clinical trials are warranted for validation of these diagnostic tools to guide second-line treatment in patients with T790M-neg NSCLC.
Conclusion
Platinum-based chemotherapy remains the recommended standard-of-care for patients with advanced EGFR mutation-positive/T790M-neg NSCLC pretreated with first- and second-generation EGFR-TKIs.19-21 However, this heterogeneous group of patients requires further evaluation to identify underlying mechanisms of resistance. The incorporation of diagnostic tools for resistance mechanisms (e.g. NGS and liquid biopsy) into prospective clinical trials is urgently needed. Immunotherapy combination therapy and other exploratory treatments may change the future treatment paradigm based on robust preclinical and clinical studies. Finally, we recommend that all T790M-neg patients be screened for clinical trials. Only well-designed and comprehensively reported prospective clinical trials can answer the questions and surmount the obstacles that we currently face.
Footnotes
Funding
The author(s) received no financial support for the research, authorship, and publication of this article.
Conflict of interest statement
B-C Liao has received honoraria/speaker fees from AstraZeneca, Roche, Boehringer Ingelheim, Merck Sharp and Dohme, Merck Serono, and Chugai. J C-H Yang received honoraria for speeches or participated in compensated advisory boards of Boehringer Ingelheim, Eli Lilly, Bayer, Roche/Genentech/Chugai, Merck Sharp and Dohme, Merck Serono, Pfizer, Novartis, Celgene, Merrimack, Yuhan Pharmaceuticals, Bristol-Myers Squibb, Ono Pharmaceutical, Daiichi Sankyo, AstraZeneca, Takeda Oncology, Blueprint Medicines, and Hansoh Pharmaceuticals. No potential conflicts of interest were disclosed by the other authors.