
Abstract
Inhibitors of fibroblast growth factor receptor (FGFR) represent an outstanding treatment approach for selected patients with urothelial cancer (UC). These agents are changing the clinical approach to a subgroup of UC, the luminal-papillary subtype, characterized by FGFR mutations, fusions, or amplification. In this review, we provide an overview of the results of recent clinical trials on FGFR tyrosine kinase inhibitors (TKIs) currently in clinical development for the treatment of UC: erdafitinib, rogaratinib, infigratinib, and the monoclonal antibody vofatamab. The Food and Drug Administration recently granted accelerated approval to erdafitinib for patients with advanced UC with alterations of FGFR2 or FGFR3 after progression on platinum-based chemotherapy. We also look at future therapeutic options of combination regimens with immune-checkpoint inhibitors as strategies for improving the antitumor effects of this class of drug, and for preventing or delaying the development of resistance.
Introduction
Bladder cancer (BC) is a common cancer worldwide whose incidence has increased in recent years. In Europe, the age-standardized incidence rate of BC is 9.0 for men and 2.2 for women. 1 BC can be divided into non-muscle-invasive (NMIBC) and muscle-invasive tumors (MIBC). Approximately 90% of all MIBC are urothelial carcinomas (UC). At diagnosis, 75% of UC are NMIBC, while 25% of cases are MIBC or metastatic disease. 2 In an estimated 5–8% of cases, UC originates in the renal pelvis or ureter (upper tract urothelial carcinoma, UTUC). 3 Patients with advanced UC are not treatable with curative intent. The first-line standard of care is cisplatin-containing chemotherapy such as gemcitabine-cisplatin or M-VAC (methotrexate, vinblastine, doxorubicin, and cisplatin), both of which are characterized by similar efficacy but with a better safety profile for the former.4,5 However, about 30% of patients are not candidates for cisplatin due to renal dysfunction, poor performance status (PS), or other comorbidities. 6 Alternative chemotherapeutic regimens, such as carboplatin-based therapies, correlate with inferior outcomes.7,8 Traditionally, the median overall survival (OS) ranged between 14 and 16 months in patients with advanced UC treated with platinum-based regimens, and long-term survival was rare.2,4
After relapse, few options are available for second-line chemotherapy. An important randomized phase III study comparing vinflunine (a third-generation vinca alkaloid) and best supportive care (BSC) with BSC alone in platinum-refractory UC reported an objective response rate (ORR) of 8.6%, a favorable safety profile, and a survival benefit for vinflunine. 9 Historically, other potential treatment options include docetaxel or paclitaxel in monotherapy.10,11
Checkpoint blockade immunotherapy recently emerged as a potentially effective treatment for these relapsed patients, with several programmed death 1 (PD-1) and programmed death–ligand 1 (PD-L1) inhibitors rapidly approved by the Food and Drug Administration (FDA), first in a postplatinum chemotherapy setting, and then as front-line treatment for cisplatin-ineligible disease.12–20 As second-line therapy, these drugs have shown an ORR of around 20%,12,13,15–17,19 without the help of predictive biomarkers approved for treating specific subsets of UC patients. Within this context, it is fundamental to find novel therapeutic agents. As part of The Cancer Genome Atlas project, the integrated genomic analysis of 131 MIBC samples revealed a high rate of somatic mutation similar to that of non-small cell lung cancer and melanoma.21,22 Robertson and colleagues proposed a new molecular classification of MIBC based on the integrated analysis of messenger ribonucleic acid (mRNA), long noncoding RNA (lncRNA), and microRNA (miRNA) expression in 412 chemotherapy-naïve samples of high grade MIBC, stratifying MIBC into five distinct subtypes: basal-squamous (35%), luminal-papillary (35%), luminal-infiltrated (19%), luminal (6%), and neuronal (5%). 23 The luminal-infiltrated subtype (19%) showed expression of CD274 (PD-L1) and cytotoxic T-lymphocyte antigen 4 (CTLA4). Initially, the luminal-infiltrated subtype, corresponding to TGCA subtype II, 21 was associated with response to atezolizumab in patients with advanced UC. 16 The basal-squamous subtype (35%) is characterized by basal keratin expression, squamous differentiation, high expression of PD-L1 and CTLA4 immune markers, and immune infiltration, making both cisplatin-based and immune checkpoint therapy valid therapeutic approaches. 17 The neuronal subtype is characterized by high expression of neuroendocrine/neuronal markers, while the luminal subtype shows high expression of luminal markers. Due to their novelty, optimal therapy for neuronal and luminal subtype has still not been defined. Recently, neuronal and luminal subtypes have been reported to be associated with better survival and response to atezolizumab than the other subtypes. 24 In UC, important developments in the molecular characterization of MIBC represent a further step forward in identifying the right treatment for the right patient. The luminal-papillary subtype is characterized by fibroblast growth factor receptor 3 (FGFR3) mutations, fusion with transforming acid coiled-coil containing protein 3 (TACC3), or amplification, suggesting that TKIs of FGFR3 may represent an interesting treatment approach for selected patients. 23 In this review, we summarize the landscape of FGFR alterations in patients with UC, the future therapeutic options, and the mechanisms of resistance to FGFR-targeted therapies.
Key genetic alterations
FGFRs (FGFR1, FGFR2, FGFR3, FGFR4) are receptor tyrosine kinases (RTKs) consisting of an extracellular ligand-binding domain [composed of three immunoglobulin (Ig)-like domains], a single-pass transmembrane domain and an intracellular tyrosine-kinase domain, that are not constitutively activated in normal cells. Upon the presence of their natural ligand, FGFRs dimerize and autophosphorylate the tyrosine residue to become activated, and phosphorylate multiple signaling proteins such as phosphatidylinositol 3 kinase (PI3K)-AKT and RAS/mitogen-activated protein kinase (MAPK), stimulating cell growth, differentiation, survival, angiogenesis, and organogenesis, depending on cell type.25–29 The FGFR family is encoded by four different genes, and alternative splicing generates two different isoforms (b/c) of the extracellular domain in each of the FGFR1, FGFR2, and FGFR3 genes, thus making seven distinct FGF receptors. 30 As these isoforms are expressed differently in various tissues and cell lines to guarantee differential roles in different tissues and cell lineages, inappropriate expression or mutation of these receptors is involved in the development of malignancies.31–33 FGFR signaling can be constitutively activated in tumor cells through amplification, missense, or fusion mutations in the coding region, and also through dysregulation of the noncoding regions, or through the alterations of epigenetic regulators or upregulation of ligands (Figure 1).34–36 Aberrations in the FGFR signaling pathway, particularly alterations in FGFR1 and FGFR3, have been shown to be involved in UC. 37
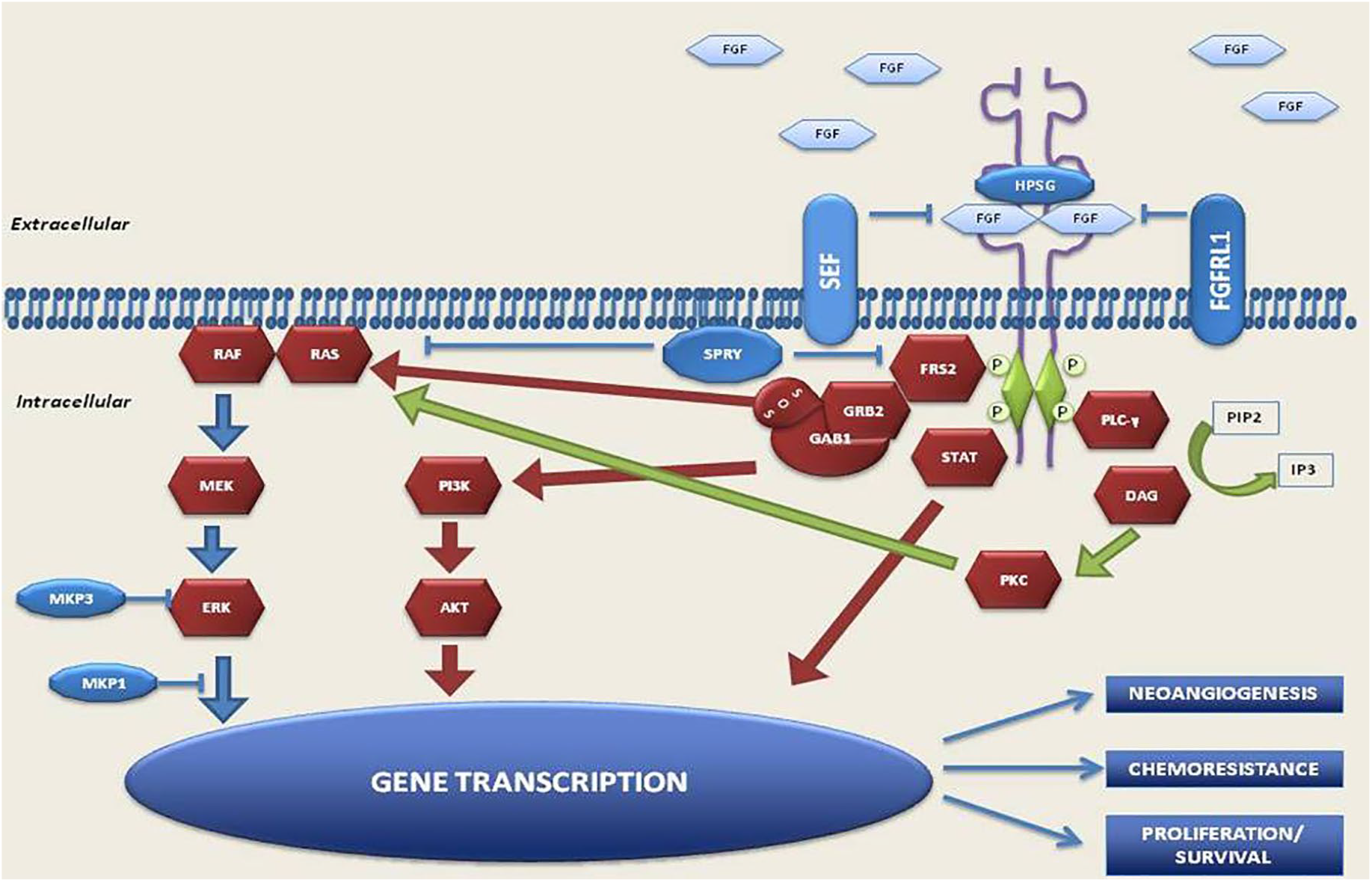
FGFR pathway.
FGFR1 amplification is present in around 42% in cancers with an FGFR alteration. 34 This gene alteration is reported in 7% of UC patients. The two splicing variants of FGFR1 (FGFR1α and FGFR1β) are expressed equivalently in normal urothelium. However, FGFR1β is the predominant form in UC, and the switch from α to β correlates with increasing tumor stage and grade. 38
The luminal-papillary subtype is characterized by FGFR3 mutations, 23 and it is interesting to note that urothelial papilloma, a benign tumor, shows frequent (75%) FGFR3 mutations. 39 Billerey and colleagues detected FGFR3 alterations in 27 (84%) out of 32 grade (G)1 NMIBCs, 16 (55%) out of 29 G2 tumors, and 5 (7%) of 71 G3 tumors, with a highly significant association (p < 0.0001) between FGFR3 mutations and low-grade disease. 40 FGFR3 mutations have also been detected in around 10% of MIBCs.41,42 The most frequent types of FGFR3 aberrations in UCs are activating mutations.21,34 FGFR3S249C, the most common aberration (21%), induces ligand-independent dimerization and activation of receptor, and is more frequently observed in low-grade UCs than in high-grade tumors. 43 Another typical alteration in UC is the fusion of FGFR3 to the TACC3 gene, leading to FGFR3 aberrant activation of downstream signaling pathways.44–46 FGFR3 amplifications are less common; however, alternative splicing of FGFR3 is implicated in the proliferation process of UC.47–49 Furthermore, despite a common histologic origin, urothelial bladder carcinoma and UTUC are two different entities with two distinct clinical pathologic profiles. A comparison between high-grade UTUC and UBC revealed that FGFR3 was more commonly altered in UTUC (35.6% versus 21.6%, respectively). 50
Molecular targeted agents in clinical trials
To date, several clinical trials are ongoing to evaluate the role of FGFR inhibitors in the treatment of UCs.
Moss and colleagues demonstrated that UTUC shows distinct mutations and different mutation frequencies compared with UBC, resulting in four different subtypes. 59 UTUCs are characterized by a higher mutation frequency of FGFR3 and lower mutation frequency of TP53 than UBCs. Dizman and colleagues found different types of FGFR3 mutations in UTUC and UBC patients. The R248C FGFR3 mutation was present in 50% of patients with UTUC compared with only 22% of UBC patients. S249C was found in 59% of UBC. The R248C mutation is induced by microsatellite instability (MSI) (mutational signature), whereas S249C mutation is induced by APOBEC. 60 Thus, the presence of a different type of FGFR mutation might explain the difference in activity. 58 However, the trial has a small sample size and results are exploratory, indicating the need for further validation.
Treatment-related adverse events
Generally, this class of drugs has manageable adverse events (AEs), as shown in Table 1. The most frequent AEs have been observed with FGFR inhibitors (erdafitinib, rogaratinib, and infigratinib), and are based on their mechanism of action. They include hyperphosphatemia, fatigue, diarrhea, and nail toxicity.67,68 The study by Siefker-Radtke and colleagues on erdafitinib did not report any treatment-related deaths. 69 In other studies on the same drug, the most common AEs included hyperphosphatemia (65%), asthenia (55%), dry mouth (45%), and nail toxicity (35%). 51 Pal and colleagues reported that the most common treatment-toxicities for patients undergoing treatment with infigratinib were hyperphosphatemia (46%), fatigue (37%), decreased appetite (32%), and stomatitis (25%). 57 Rogaratinib also has a good safety profile, Joerger and colleagues reporting hyperphosphatemia, diarrhea, and alopecia (grade 1 and 2) as the most common adverse events. 54 Retinal disorders have been observed in patients treated with FGFR inhibitors. In a phase 1 trial of rogaratinib, 3/118 patients had G1 retinal pigment epithelial detachment, while one patient experienced a G2 event consistent with serious retinopathy. 54 In a phase II trial of erdafitinib, 3% of patients discontinued treatment because of serious retinopathy. 69 Thus, an ophthalmological evaluation was implemented at baseline and during treatment in both the phase II/III study (ClinicalTrials.gov identifier: NCT03410693) of rogaratinib and in the ongoing phase III clinical trial of erdafitinib (ClinicalTrials.gov identifier: NCT03390504). All patients treated with dovitinib in the phase II trial (ClinicalTrials.gov identifier: NCT03410693) experienced at least one G4 or G4 toxicity, in particular, fatigue. One patient suffered a subdural intracranial hemorrhage, and another experienced G4 hypertriglyceridemia. 64 The development of the drug was subsequently stopped. Vofatamab was well tolerated, with few patients discontinuing treatment for AEs. The majority of AEs were G1 and G2, and included asthenia (19%), diarrhea (9.5%), flushing 14%, chills (9.5%), hypotension (9.5%), decreased appetite (19%), and increased creatinine (9.5%). Unlike FGFR inhibitors, hyperphosphatemia was not reported in patients treated with vofatamab because of the different mechanism of action of the drug. 66
Overview of benefits and AEs in clinical trial involving FGFR inhibitors in urothelial carcinoma.

AE, adverse events; FGFR, fibroblast growth factor receptor; ICIs, immune checkpoint inhibitor; ORR, overall response rate; PRs, partial responses, SD, stable disease.
Overview of ongoing trials involving FGFR inhibitors (alone or in combination) in urothelial carcinoma.

Actual enrollment.
AEs, adverse events; DLTs, dose-limiting toxicities; OS, overall survival; PD, progression disease; PFS, progression free survival; TEAEs, treatment-emergent adverse events; TESAEs, treatment-emergent serious adverse events.
FGFR inhibitors and tumor microenvironment
The tumor microenvironment (TME) consists not only of cancer cells but also of stromal and immune cells, including fibroblasts, endothelial cells, neutrophils, lymphocytes, monocytes, and macrophages.70,71 It is widely acknowledged that an active interaction between tumor cells and stromal cells is needed for tumor development, and that FGF signaling plays a key role in this network. Consequently, the action of FGFR inhibitors could also rebound on these mechanisms. There is a family of 22 FGF ligands that regulate FGFR tyrosine kinase activity in an autocrine or paracrine tissue-dependent context.72,73 For instance, FGF2 activates human dermal fibroblasts through the downregulation of the TP53 gene. In contrast, BGJ398 induces their senescence through the upregulation of TP53. 74 Myeloid-derived suppressor cells (MDSCs) are involved in the process of immune evasion and growth of the tumor through the activation of M2-type tumor-associating macrophages (M2-TAMs) and through the inhibition of CD8+ T cells and natural killer cells (NK). 75 FGFR inhibitors promote a reduction of MDSCs in the TME by targeting cytokine-producing cancer-associated fibroblasts (CAFs). 36 Migration and proliferation of endothelial cells are promoted directly through the binding of FGF2 and FGFR1, and indirectly through the induction/secretion of vascular endothelial growth factor (VEGF) from endothelial cells. 76 Huynh and colleagues recently showed that infigratinib induced vessel normalization by reducing hypoxia-inducible factor 1-alpha (HIF1α) and proangiogenic factors, hypothesizing that this could result in significant antitumor efficacy in hepatocellular carcinoma. 77 A better understanding of this autocrine and paracrine action of FGFR could help to identify an effective drug combination.
Mechanisms of resistance
There are currently no clinical reports on mechanisms of resistance against FGFR inhibitors, probably due to the novelty of FGFR as a target for TKIs. However, data on other TKIs suggest that acquired resistance is a central problem associated with FGFR inhibitors. 78–80 Treatment with FGFGR inhibitors leads to the selection of resistant cellular clones that bypass FGFR inhibition, and receive signals for cell survival and replication by activating parallel cellular signaling pathways. A recent review summarized various mechanisms of resistance to FGFR inhibitors (Figure 2), including activation of bypass signaling involving amplification or mutations in proteins appertaining to EGFR, MET, RAS, and PI3K signaling, and gatekeeper mutations conferring resistance by interfering with the binding between the receptor and the targeted agents. 81 Another important mechanism is tumor heterogeneity. Inoperable or advanced tumors have undergone clonal evolution, including FGFR-dependent clones and FGFR-independent clones, and such heterogeneity is an important obstacle to the efficacy of FGFR-targeted therapies in patients with liver cancer.82,83 UCs harboring FGFR3S249C alterations are not always sensitive to FGFR inhibitors because of their dependence on EGFR signaling rather than on FGFR3 signaling, which is repressed. Furthermore, UC cells harboring FGFR3-TACC3 fusions acquire resistance to FGFR inhibitors through the upregulation of EGFR/HER3-dependent PI3K-AKT signaling.84,85 Thus, sequencing of cell free circulating tumor (ct) DNA could be used, in addition to biopsy, to monitor the dynamic genomic landscapes of tumors, and detect the evolution of FGFR alterations. 86

Oncogenic FGFR alterations in human cancer.
Combination therapies
The benefits of FGFR inhibitors in UC have been demonstrated in numerous clinical trials. However, combination strategies involving the concomitant administration of FGFR-targeted therapies with other agents may enhance the antitumor effects of this class of drugs, and also prevent or delay the development of resistance. ICIs are monoclonal antibodies that inhibit the interaction between PD-1 and its ligand PD-L1. In a study by Powles and colleagues, PD-1/PD-L1 inhibition obtained an ORR of 52% in patients with tumor-infiltrating immune cells and high expression of PD-L1 at immunohistochemical analysis. 18 Although these results seem encouraging, the majority of patients with UC do not benefit from ICIs. In fact, treatment with ICIs has obtained ORRs ranging from 15% to 21% in platinum-refractory UC patients.12–17 There are still no valid biomarkers to predict tumor response to these treatments, but cytokines and the neutrophil-to-lymphocyte ratio look promising as prognostic and predictive indicators.87,88 However, it has been seen that the presence of an antitumor T-cell response is fundamental for the activity of immunotherapy.89–91 Recently, Sweis and colleagues, leaving from the classification of Robertson and colleagues, 23 showed that UC can be divided into T-cell-inflamed and non-T-cell-inflamed subtypes. The latter phenotype correlated with an absence of CD8+ T, and a resistance to ICIs, but was also linked to FGFR3 mutation, providing a rationale for a combination of FGFR inhibitors and anti-PD-1/PD-L1. 92 The FIERCE-22 phase II study (ClinicalTrials.gov identifier: NCT03123055) on vofatamab in combination with pembrolizumab was presented at the 2019 Annual American Society of Clinical Oncology (ASCO) meeting. The aim of the study was to prove that targeting FGFR3 (both wild type and mutant FGFR3 patients with UC), makes it possible to turn an immunologically ‘cold’ tumor into a ‘hot’ tumor. In fact, information available on other solid tumors (e.g. melanoma) suggest that the inhibition of an oncogene may induce antigen expression, decrease immunosuppressive cytokine production, and increase T cell clonality and PD-L1 expression. Luminal papillary UCs are enriched in FGFR alterations but lacking in immune infiltration. In the FIERCE-22 study, patients received one cycle of vofatamab (25 mg/kg), followed 2 weeks later by vofatamab (25 mg/kg) and pembrolizumab (200 mg) every 21 days. The ORR was 36% in the overall population, and a response was observed in both wild type (ORR 33%) and mutated (ORR 43%) FGFR3 patients. Furthermore, biopsies performed pre- and posttreatment showed that vofatamab induced immune changes, upregulating genes correlated with an inflammatory response. 93 However, data are preliminary, the duration of the response is unknown, and there are still no data on the impact of changes in the immune cell microenvironment and of PD-L1 status modifications on patient outcome. A phase Ib/II study of rogaratinib administered in combination with atezolizumab in UC is currently in progress (ClinicalTrials.gov identifier: NCT03473756). Likewise, a phase Ib-II study (ClinicalTrials.gov identifier: NCT03473743) enrolling advanced UC patients with FGFR gene alterations is ongoing to evaluate the safety and efficacy of erdafitinib plus JNJ-63723283 (an anti-PD-1). A preclinical study by Takamura and colleagues suggested that the combination of BGJ398 and the novel histone deacetylase inhibitor, OBP-801, had a synergic effect on cell growth arrest and apoptosis in high grade UC cells. 94
In TME, FGF signaling plays a pivotal role in both the survival and development of treatment resistance in tumor cells.95–97 FGFR inhibitors express their antitumor activities through direct effects on tumor cells harboring FGFR alterations, and through indirect effects on the TME, that is, regulation of angiogenesis, immune evasion, and paracrine tumor proliferation, independently of FGFR alterations. 36 Increasing our knowledge about these mechanisms is central to developing new therapeutic strategies.
Conclusion
FGFR alterations are widely present in UC, offering new opportunities for developing targeted and personalized therapies based on FGFR status. Several clinical trials are currently ongoing to demonstrate the clinical efficacy of this class of multi-targeted TKIs, and to assess the potential usefulness of FGFR alterations as a biomarker. On 12 April 2019, the FDA granted accelerated approval to erdafitinib for patients with metastatic BC harboring FGFR2 or FGFR3 alterations in progression after platinum-containing chemotherapy. 87 Further research is also warranted to investigate the combination of FGFR pathway inhibitors with other therapies to increase efficacy and overcome resistance mechanisms. Ongoing clinical trials are analyzing the combination of FGFR inhibitors and immune checkpoint inhibitors given the role that the FGF-FGFR pathway plays in TME. We believe that the FGFR pathway could open up new therapeutic avenues in this highly lethal disease, offering hope to a subset of patients by switching from cytotoxic chemotherapy to targeted agents.