
Abstract
Background:
Programmed cell death 1 (PD-1) and PD-ligand 1 (PD-L1) inhibitors represent novel therapeutic options for advanced non-small cell lung cancer (NSCLC). However, approximately 50% of patients do not benefit from therapy and experience rapid disease progression. PD-L1 expression is the only approved biomarker of benefit to anti-PD-1/PD-L1 therapy. However, its weakness has been evidenced in many studies. More recently, tumor mutational burden (TMB) has proved to be a suitable biomarker, but its calculation is difficult to obtain for all patients.
Methods:
We tested specific NSCLC genetic alterations as potential immunotherapy biomarkers. Tumor DNA was obtained from advanced NSCLC patients treated with anti-PD-1 monoclonal antibody nivolumab (n = 44) or pembrolizumab (n = 3). The mutational status of 22 genes was assessed by targeted next-generation sequencing and the association with survival was tested in uni- and multivariate models. The association between gene mutations and clinical benefit was also investigated.
Results:
The most frequently mutated genes were TP53 (49%), KRAS (43%), ERBB2 (13%), SMAD4 (13%), DDR2 (13%), STK11 (9%), ERBB4 (6%), EGFR (6%), BRAF (6%), and MET (6%). We confirmed that KRASmut patients have a better response to PD-1 inhibitors, showing a longer progression-free survival (PFS) and overall survival (OS) than KRASwt patients. In addition, we observed that patients with ERBB-family mutations, including EGFR, ERBB2, and ERBB4 all failed to respond to PD-1 antibodies, independently of KRAS status.
Conclusions:
This study suggests that the analysis of KRAS and ERBB-family gene mutational status is valuable when assessing the clinical practice for the selection of NSCLC patients to treat with PD-1 inhibitors.
Introduction
Worldwide, lung cancer is the leading cause of cancer-related death, representing a major global health concern. 1 Non-small cell lung cancer (NSCLC) accounts for 85% of lung cancer diagnoses, and approximately 50% of NSCLC patients present with stage IV disease and a 5-year survival rate of approximately 5–10%. 2 Until recently, the standard-of-care treatment for advanced NSCLC was represented by targeted therapies, when a druggable oncogenic alteration is detected, or platinum-based chemotherapy in the first-line setting and docetaxel-based chemotherapy in the following lines. 3
The recent arrival of immune checkpoint inhibitors (ICPIs) that target programmed cell death 1 (PD-1) or its ligand (PD-L1), has led to a major change in the management of metastatic NSCLC with no druggable molecular alterations. These agents are supposed to control antitumor immunity and have demonstrated unprecedented improvement in patient’s survival and disease control.4,5 Several randomized studies have demonstrated the superiority of the ICPIs (nivolumab, pembrolizumab, and atezolizumab) over docetaxel in second-line therapy for advanced NSCLC.4–6 However, the majority of advanced NSCLC fail to respond to ICPIs. 7 Thus, the identification of predictive factors to identify responder and nonresponders patients represents a large unmet clinical need.
Initially, the evaluation of PD-L1 expression on cancer cells by immunohistochemistry (IHC) was proposed as a test to predict the efficacy of anti-PD1/PD-L1 immunotherapy. 8 However, the reduced sensitivity and specificity of PD-L1 expression in predicting immunotherapy efficacy has encouraged the search for other biomarkers.9,10 Recently, a high tumor mutational burden (TMB) was found to be a positive predictive biomarker for response to immunotherapy in multiple tumor types, including lung cancer.11–13 The accumulation of mutations in tumor cells generates novel immunogenic tumor antigens (neoantigens) and consequently induces a T cell-dependent immune response against the tumor.11,12 However, the assessment of TMB by next-generation sequencing (NGS) is expensive and requires adequate pathologic material, which in advanced NSCLC is normally scarce. In addition, the use of TMB is affected by the dynamic changes that TMB can sustain during disease progression and therapies. 14
Genetic alterations in specific driver genes activate tumor cell proliferation thus supporting tumor growth. It has been demonstrated that some oncogenic pathways also influence tumor recognition by the immune system, especially T cell-mediated recognition. Smoking-associated KRAS mutations are the most frequent oncogenic alterations in NSCLC.15,16 Recent clinical evidence showed that KRAS-TP53 comutated tumors, but not KRAS-STK11 comutated tumors, have an immunogenic phenotype and are more sensitive to nivolumab.16–18
In this study, we assessed the mutational landscape of NSCLC patients treated with anti-PD-1 monoclonal antibodies pembrolizumab and nivolumab, to identify genetic alterations associated with a clinical benefit to ICPIs.
Materials and methods
Clinical samples
A retrospective consecutive series of 88 patients with locally advanced or metastatic nonsquamous NSCLC treated with ICPIs (anti-PD-1 nivolumab and pembrolizumab) were identified. Patients were treated at the Department of Oncology of Bologna, Udine and Parma (Italy), between January 2012 and December 2017. Tumor histology was confirmed using standardized diagnostic immunohistochemical workup (TTF-1, p40). After diagnostic testing, most of the residual samples were insufficient for retrospective PD-L1 IHC assessment. Demographic, clinicopathological, and outcome details for each patient were extracted from the electronic or paper medical records according to strict privacy standards. Among the total population, DNA of adequate volume and quality was available from archived formalin-fixed paraffin-embedded (FFPE) samples for 47 of the 88 patients (Table 1), whose tumors were, therefore, analyzed by target sequencing. DNA was extracted from 29 histological samples using Qiamp DNA FFPE kit (Qiagen, Venlo, Netherlands #56404) and from 18 cytological samples with Qiamp DNA Micro kit (Qiagen, Venlo, Netherlands #56304
Clinical and tumor features of NSCLC patients treated with anti-PD-1 inhibitors.

CNS, central nervous system; CR, complete response; DCB, durable clinical benefit; ECOG PS, Eastern Cooperative Oncology Group performance status; NDB, no durable benefit (SD, PR or CR < 6 months); NSCLC, non-small cell lung cancer; PD, progressive disease; (SD, PR or CR>6 months); PR, partial response; SD, stable disease.
Target sequencing
Samples were analyzed for genetic alterations using Oncomine™ Solid Tumor DNA kit (Thermo Fisher CN, Waltham, MA, USA: A26761). This panel covers more than 500 cancer-related variants in 22 genes (EGFR, ERBB2, ERBB4, MET, FGFR1, FGFR2, FGFR3, DDR2, ALK, KRAS, NRAS, PIK3CA, BRAF, PTEN MAP2K1, AKT1, TP53, STK11, CTNNB1, SMAD4, FBXW7, and NOTCH). Genomic DNA quantity and quality was assessed using Quantifiler™ Human DNA Quantification kit (Thermo Fisher, Waltham, MA, USA #4343895) for hTERT gene on Applied Biosystems™, Waltham, MA, USA 7500 Real-Time PCR System (#4351105) following the manufacturer’s instructions (PN 4344790F). Library preparation was performed according to the Oncomine™ Solid Tumor DNA kit protocol, following manufacturer’s protocol (MAN0010935). Oncomine™ Solid Tumor panel’s single pool of primers was used to perform multiplex PCR with a starting DNA quantity of 10 ng to generate 92 amplicons (115–120 bp long). Libraries were quantified using Ion Library TaqMan™ Quantification kit (Thermo Fisher, Waltham, MA, USA #4468802). After quantification, each library was diluted and pooled to obtain four equimolar library pools. Emulsion PCR and Ion Sphere Particles enrichment were performed according to the protocol Ion PGM™ Hi-Q™ View OT2 Kit (Thermo Fisher, Waltham, MA, USA #A29900; MAN0014579) using Ion OneTouch 2 system™ (#4474779). Sequencing was performed using Ion Torrent Personal Genome Machine™ (PGM™) sequencer (#4462921) with Ion PGM™ Hi-Q™ View Sequencing Kit (Thermo Fisher, Waltham, MA, USA #A30044; MAN0014583). Pools were loaded onto Ion 318 Chips. Data analysis was performed using Ion Reporter™ software selecting the AmpliSeq Colon and Lung Cancer v2 single sample Workflow. An average number of 900 variants was found for each sample. A filter chain was applied to select the variants belonging to the following types: single nucleotide variants, insertions, deletions, multiple nucleotide variants, and long deletions. A further selection was applied to remove variants below 3% of frequency, synonymous, and intronic variants. The common TP53 polymorphism (P72R) was filtered out.
Statistical analysis
Clinicopathological characteristics were summarized using descriptive analysis. Continuous variables were reported using median and interquartile ranges, and categorical variables were described using frequency distribution. The association between clinicopathological and genomic features was explored using contingency tables with the chi-squared test.
Progression-free survival (PFS) was defined as the time elapsed between immunotherapy initiation and disease progression or death from any cause, whatever occurred first. Overall survival (OS) was defined as the time elapsed between immunotherapy initiation and death from any cause or the last follow-up. Durable clinical benefit (DCB) was defined as stable disease, partial response, or complete response lasting longer than 6 months. Patients that underwent progression to disease before 6 months were classified as no durable benefit (NDB). 19 Prognostic factors in terms of OS and PFS were tested both in uni and multivariate models by Cox regression with 95% confidence interval (CI 95%). The survival curves were estimated by the Kaplan–Meier method, and the log-rank test was performed to test differences between the survival curves. The association between gene mutations and DCB/NDB was investigated with Fisher’s exact test. A value of two-sided p ⩽0.05 was considered significant. Statistical analysis was performed using STATA (StataCorp. (2015) Stata Statistical Software: Release 14.2. College Station, TX: StataCorp LP). The mutational plot was obtained using the Bioconductor package ’GenVisR’. 20 Only selected variants were plotted. When more than one mutation occurred in the same gene and patient, we plotted those with a higher frequency or higher PolyPhen2 score (suggesting damaging substitutions). Intronic mutations were evaluated using the online bioinformatic tool Human Splicing Finder. 21
Results
Patient characteristics
Demographic, clinical, and pathological features of our cohort of 47 NSCLC patients are summarized in Table 1. A total of 30 males and 17 females were included, with a mean age of 66 years at first diagnosis (range 43–85, SD ± 9.08 years). The percentage of current/former smokers is 87.23%. All patients had stage IV disease at the beginning of immunotherapy and 17% of patients had central nervous system (CNS) metastases. The majority of patients received treatment with the PD-1 inhibitors nivolumab (n = 44) or pembrolizumab (n = 3) as the second or third therapeutic line. Only one patient interrupted the therapy due to toxicity (nivolumab-related pneumonitis).
Median follow-up was 18.84 months (range 13.24–22.22 months). Median PFS and OS were 2.56 and 8.12 months, respectively. Disease progression (PD) occurred in 38 patients, and 28.95% of patients continued to receive immunotherapy beyond progression. A total of 13 patients (27.66%) had DCB, and 34 (72.34%) had NDB. A total of 33 patients (70.21%) died during the follow-up.
Mutational landscape of anti-PD-1 treatment resistance in NSCLC
NSCLC genetic alterations were assessed using NGS in 22 genes that are highly mutated in solid tumors. A median number of 2.27 nonsynonymous mutations/patient in 20 out of 22 genes were identified. All detected nonsynonymous alterations for each analyzed patient are presented in Figure 1. In addition to nonsynonymous mutations, we included as potentially pathogenic, an intronic mutation (FGFR1 - c.458-3T>G) that could affect splicing.

In our cohort (n = 47), the most mutated genes were TP53 (49%), KRAS (43%), SMAD4 (13%), DDR2 (13%), ERBB2 (13%), STK11 (9%), ERBB4 (6%), EGFR (6%), BRAF (6%), MET (6%), and other genes at a lower frequency (online supplementary Table 1). The mutational frequency distribution in KRASwt/KRASmut patients and in responders (DCB) and nonresponders (NDB) patients is shown in Figure 2.

KRAS and ERBB-family mutations are associated with outcome in NSCLC
We investigated the association between the probability of DCB and molecular characteristics (Table 2). The results highlighted a significant difference in KRAS mutation frequency between DCB/NDB groups (p = 0.012). We observed that mutations in ERBB genes (EGFR, ERBB2, ERBB4), MET, and in SMAD4 were more frequent in NDB than in the DCB group, although no significant association was found when considering each gene mutation. When gene combinations were evaluated, we observed a significant difference in the frequency of pan-ERBB, including EGFR, ERBB2, and ERBB4 mutations, between DCB/NDB groups (p = 0.009), showing a significant association with NDB status. In addition, when combining this panel with MET and SMAD4 genes, this association became even stronger.
Association between genetic alterations and clinical benefit.

DCB, durable clinical benefit (SD, PR or CR >6 months); NDB, no durable benefit (SD, PR or CR <6 months).
p value at Fisher’s exact test.
Mutually exclusive mutations.
Samples with mutations in at least one of the indicated genes.Significant p-values (<0.05) are in bold.
On univariate analysis for PFS (Table 3) patients with KRASmut tumors had a better outcome than patients with KRASwt disease (HR: 0.48, CI 95% 0.24–0.97 p = 0.041). However, patients that harbor mutations in ERBB4 and ERBB-family genes (EGFR, ERBB2, and ERBB4) were found to have a worse median PFS compared with ERBB-family wild-type patients (HR: 4.14, CI 95% 1.18–14.43 p = 0.026 and HR: 2.77, CI 95% 1.34–5.71 p = 0.006, respectively). Although a significantly worse PFS was associated with mutations in the EGFR (HR: 3.67, CI 95% 1.08–12.41 p = 0.037), NRAS (HR: 22.50, CI 95% 2.04–248.10 p = 0.011), PIK3CA (HR: 7.86, CI 95% 1.61–38.30 p = 0.011), or AKT1 (HR: 14.82, CI 95% 1.54–142.55 p = 0.011) genes, their overall mutant allele frequency was low (n = 1–3). On multivariate analysis (Table 3), the presence of KRAS (HR: 0.42, CI 95% 0.19–0.92 p = 0.033), ERBB4 (HR: 9.28, CI 95% 2.28–37.77 p = 0.002), and AKT1 (HR: 29.12, CI 95% 2.47–344.05 p = 0.007) mutation maintained their statistical significance. Moreover, a negative impact in terms of PFS was also observed by combining the ERBB-family genes with MET (HR 2.32, CI 95% 1.08–4.99 p = 0.031), SMAD4 (HR: 2.28, CI 95% 1.12–4.63 p = 0.023,) or both (HR: 2.49, CI 95% 1.23–5.04 p = 0.011).
Univariate and multivariate analysis for PFS.
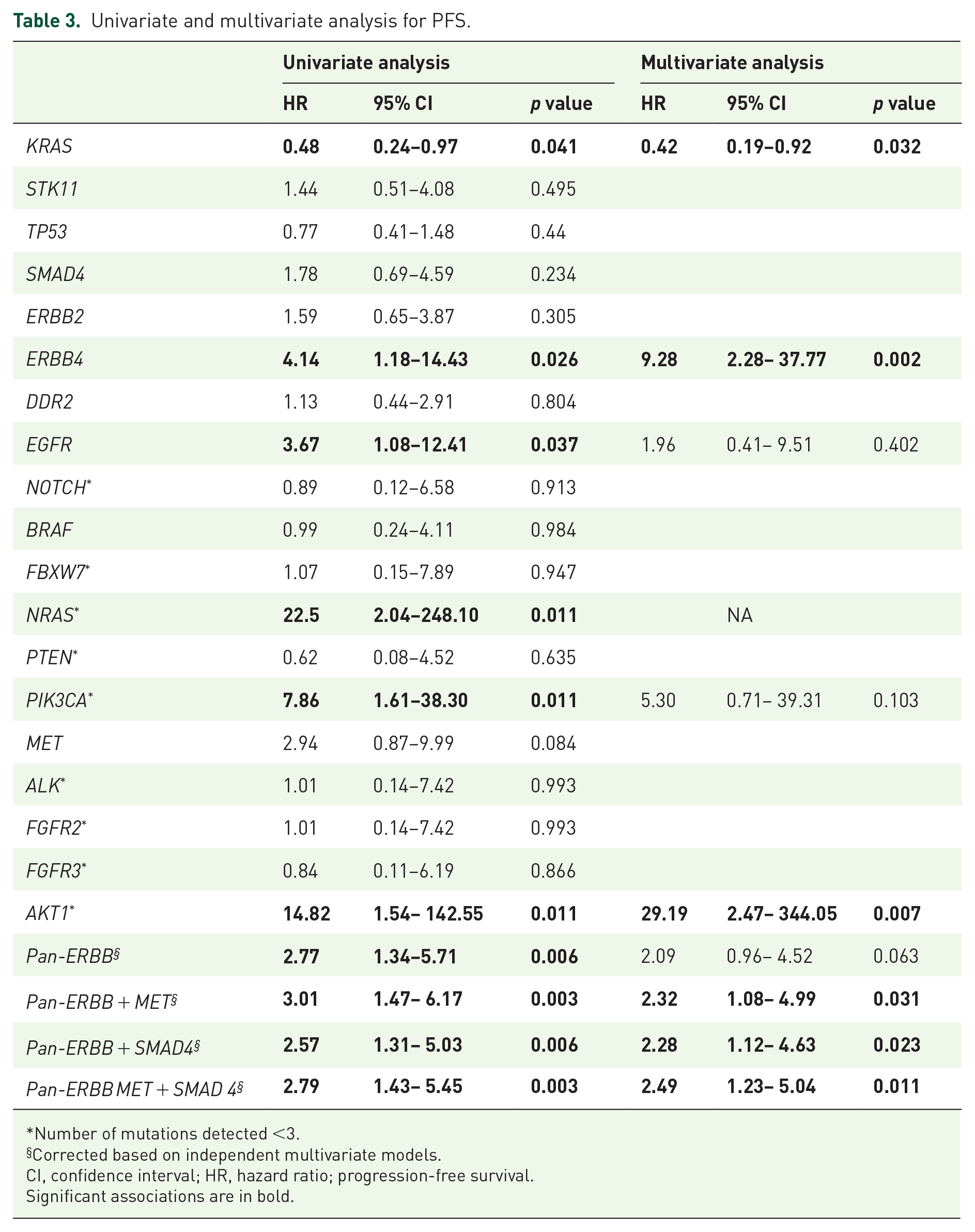
Number of mutations detected <3.
Corrected based on independent multivariate models.
CI, confidence interval; HR, hazard ratio; progression-free survival.
Significant associations are in bold.
On univariate analysis on OS, patients with KRASmut tumors had a better OS than those with KRASwt tumors (HR: 0.35, CI 95% 0.15–0.78 p = 0.010) (online supplementary Table 2). In contrast, the presence of EGFR (HR: 11.46, CI 95% 2.67–49.21 p = 0.001) or PIK3CA mutation (HR: 6.42, 95% CI: 1.35–30.52 p = 0.019), and MET alteration (HR 5.47, CI 95% 1.49–19.97 p = 0.010) were associated with worsened OS. In addition, patients that harbor mutations in ERBB-family genes were found to have a worse median OS compared with ERBB-family wild-type patients (HR: 2.19, CI 95% 1.02–4.70 p = 0.044). On multivariate analysis (online supplementary Table 2), the presence of KRAS mutation (HR: 0.39, CI 95%: 0.17–0.89 p = 0.027) and EGFR mutation (HR: 6.31, CI 95%: 1.22–32.77 p = 0.028) maintained its statistical significance, as well as the combined mutation of ERBB-family genes and SMAD4 (HR: 2.22, CI 95% 1.04–4.73 p = 0.039), or both MET and SMAD4 (HR: 2.37, CI 95% 1.11–5.03 p = 0.025).
Differences in PFS and OS were represented by Kaplan–Meier curves in Figure 3(a)–(c) and supplementary Figure 1(a)–(c), respectively.

Of note, no patient with nonsynonymous mutations in ERBB-family genes belonged to the group of DCB patients (n = 13). The 12 mutually exclusive mutations reported in these 3 genes were found to occur only in non-DCB patients, suggesting a potential implication of the ERBB-family in the mechanism of resistance to anti-PD1 blockers.
Discussion
In this study, we retrospectively analyzed by targeted sequencing a consecutive cohort of patients with locally advanced or metastatic nonsquamous NSCLC, who were treated with the anti-PD1 inhibitors nivolumab or pembrolizumab. The aim was to highlight some of the genetic determinants of anti-PD-1 resistance.
The arrival of ICPIs, targeting PD-1 and its ligand PD-L1, has led to a major change in the treatment of metastatic nononcogene addicted NSCLC, with a significant improvement of survival and disease control. The anti-PD-1 and anti-PD-L1 inhibitors are currently the treatment of choice after first-line chemotherapy (CT). Specifically, the anti-PD-1 nivolumab and anti-PD-L1 atezolizumab are approved as second and later therapeutic lines for advanced NSCLC, regardless of PD-L1 expression. Pembrolizumab has been approved as a first-line monotherapy for patients with advanced NSCLC and PD-L1 score ⩾50%, 9 and is the standard treatment for NSCLC patients who progressed to first-line CT with PD-L1 score ⩾1%. 5 Recently, positive results from several clinical trials led to the marketing approval of a first-line combination of pembrolizumab plus CT for patients with advanced NSCLC,22–24 and atezolizumab plus bevacizumab plus CT as first-line treatment for advanced nonsquamous NSCLC. 25 In addition, nivolumab plus ipilimumab represents another promising therapeutic options for patients with untreated, advanced NSCLC with high TMB. 26
Despite unprecedented improvement in outcomes with the use of anti-PD-1 and anti-PD-L1 inhibitors, the majority of NSCLC patients fail to respond to ICPIs. Thus, the identification of predictive clinical factors and biomarkers of clinical response or resistance to this therapy in metastatic NSCLC represents a significant need for the appropriate selection of responders from nonresponders.
The extreme heterogeneity characterizing advanced nonsquamous NSCLC represents a clinical challenge and affects the development of effective therapeutic strategies for patients with such diagnosis. Simultaneously, the high incidence of KRAS mutations and the lack of specific and effective agents targeting KRAS have led to a growing interest in KRAS-mutated NSCLC.
Similar to previous reports,4,6,27 we observed that patients with KRAS mutations benefit most from PD-1 blockade. Indeed, we found that KRAS-mutant patients had statistically significant longer OS and PFS as compared with patients with KRAS wild-type disease. Furthermore, we also showed that the presence of nonsynonymous KRAS mutations is associated with a DCB. The mechanisms underlying this higher sensitivity to immunotherapy of KRAS-mutant tumors are still under investigation. 28 It is known that KRAS mutations are generally associated with smoking and high TMB which, in turn, results in the generation of immunogenic neoantigens that could stimulate immune response. 29
The most innovative finding of our analysis is the strong association between HER/ERBB pathway mutations and the lack of response to anti-PD-1 inhibitors. Previous reports suggested using EGFR mutations as biomarkers of resistance to ICIs.4,27,30,31 In our cohort, the patients harboring mutations in EGFR were found to have NDB from immunotherapy.
When we analyzed all ERBB-family genes contained in the NGS panel (EGFR, ERBB2, and ERBB4), we obtained evidence of a statistically significant negative impact of ERBB-family mutations on patient’s outcomes. Of note, we found that patients with mutations in ERBB pathway genes had worse PFS and OS than patients with ERBB-family wild-type disease. Our findings suggest that ICPIs with anti-PD-1 may not be effective in patients with EGFR or any ERBB-gene mutant nonsquamous NSCLC.
The HER/ERBB pathway is frequently mutated in NSCLC and has been linked to PD-L1 upregulation and reduced antigen presentation to CD8+ T cells. 32 Specifically, EGFR and ERBB2 pathway activation was shown to be responsible for PD-L1 increased expression on the surface of lung cancer cells 33 and for the impairment in MHC class I antigen presentation.34,35 In addition, EGFR-mutant patients have a high frequency of inactive tumor infiltrating lymphocytes. 36 These mechanistic studies provide a biological explanation of the results we obtained in the clinic.
Recently, it was also demonstrated that KRAS-mutant NSCLC include different subtypes with different biology, prognosis, and response to ICPIs.16,17 Specifically, Skoulidis and colleagues proved that alterations in STK11 are associated with de novo resistance to anti-PD-1 therapy despite the presence of intermediate/high TMB. This is partially due to the reported association between lack of PD-L1 expression and STK11 inactivation. In addition, KRAS/TP53 comutation was reported to be associated with increased TMB and PD-L1 expression 16 and a more favorable response to anti-PD-1 blockade. 31 In our series, we observed nine (45%) KRAS-mutant tumors with intact STK11/LKB1 and TP53 (KO or K only), nine KRAS-mutant harboring mutations in TP53 (KP), and two (10%) KRAS-mutant bearing mutations in STK11/LKB1 (KL). No triple-mutant (KRAS/ TP53/ STK11) tumors were detected. These frequencies were different from the ones reported in the Skoulidis study. Unfortunately, the small number of cases belonging to the KO, KP and especially KL subgroups prevented us from verifying the association between KRAS comutations and outcome. However, it is worth noting that of the four patients with STK11 mutations, one had a durable benefit lasting longer than 6 months. On the bases of these results, we cannot draw any definitive conclusion about the impact of these alterations on PFS and OS of our population. It is likely that the type of molecular assay used (NGS panel versus WES) may account for the different findings.
Finally, we assessed the impact on prognosis of other mutations such as SMAD4, NRAS, PIK3CA, AKT, and MET mutation. SMAD4 and MET mutations were correlated with worse outcomes on univariate analysis in our series. However, larger prospective studies are required to confirm these findings.
Our study has some limitations, including the retrospective observational design and the small sample size. However, it provides a real-world scenario of genomic analysis in a consecutive series of advanced nonsquamous NSCLC patients receiving immunotherapy with anti-PD-1 in three large Italian hospitals, in the context of standard clinical practice, using a commercially available NGS panel.
Conclusion
Our findings suggest that the presence of KRAS mutations and the absence of ERBB-family gene mutations should be further evaluated as biomarkers of benefit to nivolumab and pembrolizumab treatments.
Supplemental Material
Supplementary_Figure_1 – Supplemental material for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC
Supplemental material, Supplementary_Figure_1 for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC by Marika Cinausero, Noemi Laprovitera, Giovanna De Maglio, Lorenzo Gerratana, Mattia Riefolo, Marianna Macerelli, Michelangelo Fiorentino, Elisa Porcellini, Vanessa Buoro, Francesco Gelsomino, Anna Squadrilli, Gianpiero Fasola, Massimo Negrini, Marcello Tiseo, Manuela Ferracin and Andrea Ardizzoni in Therapeutic Advances in Medical Oncology
Supplemental Material
Supplementary_table_1 – Supplemental material for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC
Supplemental material, Supplementary_table_1 for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC by Marika Cinausero, Noemi Laprovitera, Giovanna De Maglio, Lorenzo Gerratana, Mattia Riefolo, Marianna Macerelli, Michelangelo Fiorentino, Elisa Porcellini, Vanessa Buoro, Francesco Gelsomino, Anna Squadrilli, Gianpiero Fasola, Massimo Negrini, Marcello Tiseo, Manuela Ferracin and Andrea Ardizzoni in Therapeutic Advances in Medical Oncology
Supplemental Material
Supplementary_table_2 – Supplemental material for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC
Supplemental material, Supplementary_table_2 for KRAS and ERBB-family genetic alterations affect response to PD-1 inhibitors in metastatic nonsquamous NSCLC by Marika Cinausero, Noemi Laprovitera, Giovanna De Maglio, Lorenzo Gerratana, Mattia Riefolo, Marianna Macerelli, Michelangelo Fiorentino, Elisa Porcellini, Vanessa Buoro, Francesco Gelsomino, Anna Squadrilli, Gianpiero Fasola, Massimo Negrini, Marcello Tiseo, Manuela Ferracin and Andrea Ardizzoni in Therapeutic Advances in Medical Oncology
Footnotes
Author contributions
AA designed the study; MC, MT, MM, VB, AS, GF, and FG selected patients and collected clinical information; MF, NL, and EP performed NGS experiments and analyzed results; LG and MR performed statistical analyses; MFi and GDM performed the histo-pathological examinations; MN contributed to data interpretation; AA, MF, MC, NL, and LG interpreted and discussed the data and wrote the manuscript. All authors revised and approved the manuscript.
Conflict of interest statement
AA reports grants and personal fees from BMS, personal fees from MSD, personal fees from Eli-Lilly, personal fees from Boehringer, personal fees from Pfizer, and grants from Celgene outside the submitted work. MT reports advisory boards and/or speaker’s fee for BMS and MSD. The other authors declare that they have no conflicting interests.
Ethics approval and consent to participate
The study was approved by Comitato Etico Unico Regionale (C.E.U.R.; approval ID: CEUR-2017-0s-123-ASUIUD), Friuli-Venezia Giulia Region. Before study entry, all patients provided written and voluntary informed consent for inclusion, collection and use of clinicopathological data and samples.
Funding
The research leading to these results has received funding from AIRC IG grant - ID. 19026 project – P.I. AA. MF lab is supported by AIRC and Fondazione Pallotti funds. MR is a Fondazione Famiglia Parmiani fellow.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.