
Abstract
Renal transplantation has become the sole most preferred therapy modality for end-stage renal disease patients. The growing tendency for renal transplants, and prolonged survival of renal recipients, have resulted in a certain number of post-transplant colorectal cancer patients. Antitumor pharmacotherapy in these patients is a dilemma. Substantial impediments such as carcinogenesis of immunosuppressive drugs (ISDs), drug interaction between ISDs and anticancer drugs, and toxicity of anticancer drugs exist. However, experience of antitumor pharmacotherapy in these patients is limited, and the potential risks and benefits have not been reviewed systematically. This review evaluates the potential impediments, summarizes current experience, and provides potential antitumor strategies, including adjuvant, palliative, and subsequent regimens. Moreover, special pharmaceutical care, such as ISDs therapeutic drug monitoring, metabolic enzymes genotype, and drug interaction, are also highlighted.
Introduction
Renal transplant, commonly performed for end-stage renal disease (ESRD), is an alternative to dialysis. After renal transplant, immunosuppressive drugs (ISDs) are used continuously to prevent allograft rejection. Improvements in ISDs have led to significantly enhanced graft survival. As a result, kidney transplant recipients (KTRs) have greater accumulation and longer exposure to ISDs than ever before. Concomitantly, the incidence of severe side effects, such as malignancies, has increased, which has become a major obstacle for long term survival in KTRs. 1
Colorectal cancer (CRC) is the third most common cancer worldwide, and the second highest cause of cancer mortality. It has been reported that the risk of CRC is higher in KTRs than in the nontransplant population. 2 Antitumor therapy, especially antitumor pharmacotherapy, can significantly improve the prognosis of CRC. However, substantial impediments such as carcinogenesis of ISDs, drug interaction between ISDs and anticancer drugs, and toxicity of anticancer drugs exist. In this review, we systematically evaluate the potential risks of antitumor drugs in order to provided optimal antitumor pharmacotherapy in KTRs with CRC.
Epidemiology of CRC after renal transplant
Malignancy is one of the complications most often encountered in the later period post transplantation. Well-recognized types of malignancies are skin, kidney, lung, bladder, liver, lymphoma, Kaposi’s sarcoma, and carcinoma of the vulva perineum. 3 It is generally recognized that renal transplant has also been related to increased risk of CRC,4,5 although the risk increasing of rectal cancer is much more controversial. 6 Previously reported incidence of CRC in KTRs ranges dramatically from 0.02% to 3.62%2, 3, 7–14, 2, 6, 15–38 (Table 1). This variation might be due to differences in geographical location, duration and degree of immunosuppression, age of renal transplant, complication, etc. Anyway, the growing tendency for renal transplant and prolonged survival of KTRs have resulted in a certain number of CRC patients after renal transplant.
The incidence of colorectal cancer in renal transplant recipients.

CRC, colorectal cancer.
CRC in KTRs has a worse 5-year survival rate than in the general population.2,23 Papaconstantinou and colleagues researched patients in Israel Penn International Transplant Tumor Registry and found that transplantation patients had a younger mean age at colorectal cancer diagnosis (58 versus 70 years; p < 0.001), and a worse 5-year survival (overall, 44% versus 62%, p < 0.001; Dukes A and B, 74% versus 90%, p < 0.001; Dukes C, 20% versus 66%, p < 0.001; and Dukes D, 0% versus 9%, p = 0.08). 2 Kim and colleagues investigated the CRC in KTRs in Korea from 1994 to 2007, and found that recurrence (35.2% versus 15.2%, p = 0.048) and 5-year survival (40.2% versus 67.8%; p = 0.044) were worse in the transplant group than in the nontransplant group. 23 This difference was more significant in advanced cancers. The 2-year survival of the transplant group was significantly worse than the nontransplant group with advanced cancer (stages III–IV; 45.7% versus 71.6%; p = 0.023).
Besides immunosuppression, the decreased survival is also due to inadequate anticancer treatment. 23 Surgical resection and radiotherapy remain the preferred treatment for most early-stage and locally invasive CRC in KTRs. Pharmacotherapy is necessary to prevent tumor relapse and development of metastases and achieve adequate palliation. However, pharmacotherapy of CRC in KTRs is a dilemma, and has significant impediments.
Immunosuppressive therapy in KTRs with CRC
ISD selection in KTRs with CRC
Induction period of immunosuppressive therapy starts before, or at the time of, kidney transplantation. Hereafter, initial maintenance therapy initiates and a combination of ISDs is recommended. If there is no acute rejection, lowest planned doses of maintenance ISDs are used as long-term maintenance therapy starting 2–4 months after transplantation. The mean onset time of CRC is 10.4 years after transplantation, 39 which is the long-term maintenance period of immunosuppressive therapy. In this period, the combination of ISDs with different mechanisms of action and at reduced doses has recommended by most society- and government-sponsored guidelines. Conventional maintenance regimens includes a calcineurin inhibitor (CNI) and an antiproliferative agent, with or without corticosteroids. This strategy minimizes morbidity and mortality associated with each class of agent while maximizing overall effectiveness.
Carcinogenesis is an important consideration for choosing of ISD in the maintenance period. Calcineurin controls the nuclear factor of activated T cells (NFAT), an essential transcriptional factor for the activation of T lymphocytes, 40 and is, therefore, a target of cyclosporine A (CsA) and tacrolimus (TAC). The overexpression of calcineurin in human colorectal adenocarcinomas, and the activation of NFAT in human colon cancer cell lines, have been reported. 41 CsA and TAC were shown to reduced cell growth in colony cell lines (HT29). 41 Paradoxically, some research also showed that CsA and TAC increased transforming growth factor-beta, an effector clearly associated with tumor growth.42,43 The controversy surrounding the anti-CRC activity of CNI is still unresolved, but TAC is associated with decreased acute rejection rates and is generally better tolerated than CsA. 44 Thus, we suggest the administration of TAC rather than CsA.
Azathioprine (AZA), sirolimus (SIR), 45 and mycophenolate mofetil (MMF) 46 are commonly used antiproliferative agents. The use of AZA has been associated with neoplastic development post-transplantation, particularly an increased risk of cutaneous squamous cell carcinomas. The mechanism of action is postulated to be the inhibition of repair splicing and induction of codon misreads via intercalation of DNA . 47 AZA is not preferred because of its inferior ability to prevent acute rejection and its poorer side effect profile than MMF. SIR, an inhibitor of the mammalian target of rapamycin (mTOR), can suppress the growth of tumors. 45 A meta-analysis of 21 randomized trials showed that SIR was associated with a 40% reduction in the risk of malignancy (adjusted HR 0.60, 95% CI 0.39–0.93, p = 0.02), but 43% increased risk of death (adjusted HR 1.43, 1.21 to 1.71; p < 0.001) compared with other ISD. 46 Elevation of inosine monophosphate dehydrogenase was found in some solid tumors. MMF blocked purine biosynthesis via inhibition of inosine monophosphate dehydrogenase.18,48 Some population studies have suggested that the risk of developing a malignancy is not higher with MMF, and may actually be associated with a decreased risk.18,48,49 It is still not clear whether SIR- or MMF-containing regimens improve patient outcomes. SIR showed no particular superiority to MMF, but was associated with increased risk of graft loss when combined with CNI, even when combined with a reduced dose of CNI. Hence, we suggest administration of MMF rather than SIR.
Kidney Disease: Improving Global Outcomes (KDIGO) guidelines suggested that, among patients at low immunologic risk and who receive induction therapy, prednisone should be discontinued during the first week after transplantation. This is based on the desire to minimize long-term glucocorticoid exposure, thereby decreasing the risk of adverse effects with this agent. However, many transplant centers continue low-dose glucocorticoid therapy in all patients, regardless of the risk of acute rejection.
Future perspectives and clinical relevance of ISD
The controversy surrounding the anti-CRC activity of CNI is still unresolved, despite its ability to act both as a tumor suppressor and as an oncogenic activity inducer in CRC. CNI dose may be an important factor in its immunosuppressive and anticancer effects. In a phase I/II trial on 44 patients with advanced non-small cell lung carcinoma, low-dose CsA (1–2 mg/kg per day) was compared with high-dose CsA (3–6 mg/kg per day) in association with etoposide and cisplatine. In this small series, the authors reported a significant increase in survival of patients treated with low-dose CsA, with a 2-year survival of 25% compared with 4% with high-dose CsA. Kaplan–Meier survival curves were significantly different for these two groups by log-rank test (p = 0.047). 50 In this regard, more studies are needed to evaluate the effects of different doses of CNIs on CRC and immunology.
NFAT is a family of common cellular transcription factors that is initially identified in T cells. The NFAT family includes five members: NFAT1, NFAT2, NFAT3, NFAT4, and NFAT5. Previous research showed that NFATc2 is overexpressed in CRC tissues compared with normal tissue margins (p ⩽ 0.05), and might be used as a therapeutic target. 51 The study of Vaeth and colleagues also showed that selective NFAT targeting (NFAT1, NFAT2, or a combination of both) in T cells might ameliorates graft-versus-host disease while maintaining antitumor activity. 40 Hence, further investigations should identify specific targets for calcineurin or NFAT to anti-tumor. More importantly, selective CNIs or natural compounds against targets could be useful clinically for treating CRC in KTRs.
Pharmacotherapy of CRC
For CRC, the available agents include oxaliplatin, irinotecan, parenteral and oral fluoropyrimidines, anti-angiogenesis drugs, anti-epidermal growth factor receptor (EGFR) monoclonal antibody (MoAb), and drugs targeting the programmed cell death-1 (PD-1)/programmed death-ligand 1 (PD-L1). 52
For patients with indications for adjuvant chemotherapy, a combination chemotherapy of 5-fluorouracil (5-FU)/leucovorin (LV)/oxaliplatin (FOLFOX) or capicitabine/oxaliplatin (CapeOx) is the preferred option, while fluoropyrimidine-based regimen (5-FU/LV or capecitabine alone) is recommended for patients who cannot tolerate combined chemotherapy. 53 5-FU/LV/irinotecan (FOLFIRI) and 5-FU/LV/oxaliplatin/irinotecan (FOLFOXIRI), as well as the regimens mentioned above, are recommended as palliative regimens. Bevacizumab is recommended to add to chemotherapy in palliative therapy. Cetuximab or panitumumab is superior to bevacizumab for patients with a wild-type KRAS gene.54,55 Some new drugs, such as aflibercept, regorafenib, trifluridine-tipiracil, S-1, tegafur-uracil, nivolumab, and pembrolizumab, have been suggested in subsequent therapy . 56 For KTRs with CRC, anticancer regimens should be optimized as far as possible to avoid interfering with immunosuppressive therapy.
Possible interaction between ISD and anticancer drugs
Whenever patients take more than one medication, they are at risk of a drug interaction. For post-transplantation CRC patients, potential interaction between ISDs and anticancer drugs must be checked before it occurs. Drug interactions occur in two different ways: pharmacokinetic interaction and pharmacodynamic interaction.
Pharmacokinetic interaction
Pharmacokinetic interaction may occur if one drug affects another drug’s absorption, distribution, metabolism, or excretion. There is no research on pharmacokinetic interactions between ISDs and antitumor drugs, although some interaction in absorption, distribution, metabolism, and excretion could be predicted.
Interaction in drug absorption
ISDs are absorbed in the gastrointestinal tract, and bioavailability varies from 14% to 94% (Table 2). Many factors can affect the absorption of ISDs; for example, colitis can affect the secretion and excretion of bile, which disturbs the enterohepatic circulation and bioavailability of MMF. Anticancer drugs have gastrointestinal toxicity, leading to diarrhea, vomiting, and constipation, which disturbs gastrointestinal function and subsequently ISDs absorption.
The absorption and distribution of immunosuppressive agents and anticancer drugs.

Diarrhea
Diarrhea is most commonly described with chemotherapeutic drugs such as fluoropyrimidines and irinotecan. Irinotecan is associated with both early- and late-onset diarrhea. Early-onset diarrhea occurs within several hours of drug infusion and has an incidence of 45–50%. It is cholinergically mediated and usually well controlled by subcutaneous or intravenous atropine. Late-onset diarrhea induced by irinotecan is not cholinergically mediated, and is unpredictable. The median time to onset is approximately 6 days, with a 350 mg/m2 every 3-week schedule, and 11 days with a weekly schedule (125 mg/m2). 57 The pathophysiology of late diarrhea appears to be multifactorial, with some association with the polymorphism of UGT1A1. 58 In some studies, homozygotes for the UGT1A1*28 (and, to a lesser degree, heterozygotes) have had significantly higher rates of diarrhea with irinotecan. The UGT1A1 genotype should be detected before the initiation of irinotecan-containing regimens such as FOLFIRI and FOLFOXIRI. It is advised that the starting dose of irinotecan should be lowered in patients known to be homozygous for UGT1A1*28.
Diarrhea can occur in all schedules with fluoropyrimidines. A meta-analysis showed that more frequent grade 3/4 diarrhea could be found in capecitabine-based groups (507/2658) than in 5-FU-based groups (333/2624). 59 The pathophysiology of diarrhea appears to be multifactorial, with some association with the polymorphisms of dihydropyrimidine dehydrogenase (DPD) and thymidylate synthetase (TYMS).60,61 Polymorphisms of DPD and TYMs lead to deficient enzyme activity and inadequate degradation of fluoropyrimidines, and, thus, subsequently increase the risk of severe toxicity such as diarrhea. Testing of DPD and TYMS polymorphism for fluoropyrimidine-based chemotherapy is a controversial area nowadays, 60 and has not been widely adopted. We advise detecting DPD and TYMS genotype in KTRs to identify high-risk patients, and making an early decision to reduce dose or select an alternative treatment regimen.
In contrast to fluoropyrimidines and irinotecan, anti-EGFR MoAbs related diarrhea is generally not pervasive or severe.62,63 But adding anti-EGFR MoAbs to standard chemotherapy increased the rates of diarrhea. In a meta-analysis of 18 randomized controlled clinical trials, the incidence of grade 3–4 diarrhea was 18% (95% CI 14–23%) in the anti-EGFR MoAbs arm and 11% (95% CI 8–14%) in the control arm. 64 Using a fixed-effects model, the overall result showed that anti-EGFR regimens are associated with a significantly higher risk of severe diarrhea (RR of 1.66, 95% CI1.52–1.80). 64 Other studies also showed similar results.46,65
Considering the cornerstone status of fluoropyrimidines in the treatment of CRC, fluoropyrimidine monotherapy or combination chemotherapy still applies to KTRs with CRC, although they have a high risk of diarrhea. DPD and TYMS genotype test might be helpful in these patients. Regimens containing fluoropyrimidines and irinotecan (FOLFIRI and FOLFOXIRI) are not preferred, but can be used in oxaliplatin-refractory patients. UGT1A1 genotype should be detected before the initiation of irinotecan-based regimens. Triple combinations of fluoropyrimidines, irinotecan and anti-EGFR MoAbs (cetuximab/panitumumab + FOLFIRI) are less recommended due to their synergistic effect of diarrhea. For patients needing intensive treatment, the triple combination of fluoropyrimidines, oxaliplatin, and anti-EGFR MoAbs (cetuximab/panitumumab + FOLFOX or cetuximab/panitumumab + CapeOx) might be a choice, but also increase the risk of other adverse drug reactions (ADRs) besides diarrhea.
Anticancer regimens optimization can minimize the risk of diarrhea, but cannot avoid diarrhea completely. It has been confirmed that the absorption of MMF66,67 and CsA 68 is little affected by diarrhea, and needs no further dose adjustments. However, severe diarrhea (more than three loose stools daily) substantially increased with exposure to TAC, which is also reflected as an elevation of the trough level. Therefore, dose adjustments of TAC in patients with severe diarrhea must be monitored carefully, especially when doses of MMF are also reduced. 68 Various explanations for the effect of severe diarrhea upon exposure to TAC have been proposed, but the most important one is enhanced absorption as a consequence of a damaged intestinal barrier and reduced intestinal metabolism. 69 It is generally considered that mild diarrhea might be an effect of exposure to TAC, but probably to a lesser extent. 69 However, in the study of van Boekel and colleagues, 69 mild diarrhea did not affect the exposure and trough level of TAC, and thus showed no evidence for the presence of hidden TAC overexposure in patients with mild diarrhea while on treatment with TAC and MMF.
Emesis and constipation
Trifluridine/tipiracil has moderate to high emetic risk, which means more than 30% frequency of emesis. Oxaliplatin and irinotecan have moderate emetic risk, which means a 30–90% frequency of emesis. The emetic risk of other anticancer drugs used in CRC is low or minimal. Preventive antiemetic therapies were used widely before chemotherapy and can prevent most of the emesis. Additional antiemetic therapy will be given when patients have breakthrough nausea. Dosing interval should be adjusted in order to minimize the risk of vomiting with ISDs. The Tmax values of TAC, CsA, SIR, and MMF are 1–3 h, 1.5–2.0 h, 1.0 h, and 0.9–1.8 h, respectively. Hence, ISDs should be taken orally 1–3 h before the beginning of an anticancer regimen.
The effects of constipation on ISDs exposure have not been reported. Anticancer drugs rarely cause constipation directly. Antiemetic therapies, such as 5-hydroxytryptamine receptor antagonists, often induce constipation that slows intestinal transit time. The initial management of constipation includes patient education, dietary changes, bulk-forming laxatives, or the use of nonbulk-forming laxatives or enemas. Considering multiple concurrent drugs, it is preferred for KTRs with CRC to increase fluid and fiber intake to prevent constipation. Laxatives are used only for existing constipation, because laxatives affect CNI absorption. 70
Attention should be paid to the interaction between ISD and proton pump inhibitors (PPIs) as it is widely implicated in chemotherapy-induced gastrointestinal dysfunction. It has been confirmed that PPIs reduce absorption of MMF by elevating gastric pH value and decreasing dissolution of MMF.71,72 Rupprecht and colleagues demonstrated that the maximum concentration of a single dose of MMF was reduced by 57% in healthy volunteers, and exposure was reduced by 27% when given 1 h after a single dose of pantoprazole 40 mg. 73 A number of studies designed to evaluate the impact of MPA pharmacokinetics in transplant recipients on various PPI therapies have shown similar effects on MMF exposure, but failed to demonstrate a significant impact on rejection rates, graft function, or graft survival. 74 However, physicians should pay more attention to monitoring MMF levels in the presence of PPIs.
The presence of clinically relevant pharmacokinetic drug interaction between PPIs and TAC remains a matter of controversy. 75 In vivo studies using human liver microsomes have shown that omeprazole inhibits CYP3A4-mediated metabolism of TAC competitively. 75 In contrast, Pascual and colleagues estimated the potential interaction between omeprazole and TAC in renal transplant recipients and concluded an absence of important drug interaction. 76 In these conditions, rabeprazole or H2RA were proposed as a safer treatment option than omeprazole in KTRs receiving TAC.
Interaction in drug distribution
When a drug is displaced from its plasma binding protein, increased unbound drug concentrations theoretically cause an increase in drug effect, with potentially toxic results. 77 However, some scholars have presented theoretical arguments from a few cases where protein-binding changes were clinically significant. Benet and colleagues considered that changes in plasma protein binding have little clinical relevance. 77 We also summarize the distribution and plasma protein binding of ISD (Table 2) in order to minimize the potential risk of interaction in drug distribution. Methylprednisolone, TAC, SIR, and MMF bind mainly to albumin, and prednison binds mainly to corticosteroid transporters, while CsA binds mainly to lipoprotein. Plasma protein binding rates of ISDs vary from 40% to 99%. TAC, MMF, and oxaliplatin bind mainly to albumin, but the specific binding site and binding pattern to albumin are still unknown. On the other hand, it is still unclear what proportion of plasma protein is bound to each drug. So it is uncertain whether oxaliplatin competes for protein binding with TAC and MMF. Therapeutic drug monitoring of ISD cannot predict protein binding competition because it represents the total plasma concentration not the free concentration. But we also recommend therapeutic drug monitoring of ISDs because no other indicator can be used in this condition.
Interaction in drug metabolism
The metabolism of ISD and anticancer drugs are summarized in Table 3. In humans, three carboxylesteras (CES) have been identified: human liver CES (CES1), human intestinal CES (CES2), and human brain CES (CES3). 78 MMF is an inactive ester prodrug, and undergoes hydrolysis to form an active drug, mycophenolic acid. Hydrolysis occurs in the intestine, plasma, and liver. Liver hydrolysis by CES1 has been demonstrated to be the most efficient pathway. 79 Capecitabine, a prodrug of 5-fluorouracil, is first metabolized to 5′-deoxy-5-fluorocytidine (5′-DFCR), mainly by CES2. 80 Hence, the combination of MMF and capecitabine rarely causes competition of CES.
The metabolism and excretion of immunosuppressive agents and anticancer drugs.

5-FU, 5-fluorouracil; CES, carboxylesterases; CYP, cytochrome P450; GMPS, Guanosine monophosphate synthetase; HGPRT, Hypoxanthine-guanine-phosphoribosyl-transferase; IMPD, Inosine monophosphate dehydrogenase; P-gp, P-glycoprotein; TPMT, thiopurine S-methyltransferase; UGT, UDP-glucuronosyltransferase; XO, Xanthine oxidase.
Irinotecan is a prodrug whose hydrolysis results in the formation of the active metabolite, 7-ethyl-10-hydroxy camptothecin (SN-38). 81 Both CES1 and CES2 appear to contribute to the hydrolysis of irinotecan to SN-38. 82 Drug activation in the intestine and kidney are likely major contributors to SN-38 production in vivo. Studies have also shown that CES2 is commonly expressed in tumor tissue and is correlated with activation of irinotecan. 83 As MMF is hydrolyzed mainly by liver CES1, competition with irinotecan for CES seems unlikely.
CYP3A4 activates regorafenib and prednisone, but inactivates CsA, TAC, and SIR. Competition of CYP3A4 between regorafenib and ISD has not been reported, but can be anticipated. Competition of CYP3A4 might increase the plasma concentration of regorafenib and decreased the plasma concentrations of the active metabolites M-2 and M-5, and may lead to decreased efficacy and toxicity. The plasma concentration and immunosuppressive effect of CsA, TAC, and SIR might be increased, while that of prednisone might be decreased when competition of CYP3A4 occurs. As CNIs such as CsA or TAC are necessary for maintenance immunosuppressive therapy, the use of regorafenib should be avoided as far as possible.
The active metabolites of irinotecan (SN-38) and MMF (mycophenolic acid) are inactivated by uridine diphospho-glucuronosyltransferase 1A (UGT1A). The combination of irinotecan and MMF has the potential for competition with UGT1A, although this has not yet been confirmed. The potential competition might lead to the accumulation of the active metabolites SN-38, which significantly enhance the possibility of fatal late diarrhea. 58
Regorafenib has proved to be a UGT1A inhibitor. 84 Inhibition of UGT1A by regorafenib or competition of UGT1A by irinotecan might result in the accumulation of mycophenolic acid, and thus reinforce the immunosuppressive effect. Considering the wide use of MMF in immunosuppressive therapy, the use of irinotecan and regorafenib is restricted for these patients.
Interaction in drug excretion
The excretion of ISD and anticancer drugs are summarized in Table 3. Capecitabine, oxaliplatin, prednisone, and MMF are excreted in the urine without the participation of a carrier. So the probability of excretive interaction is small.
Pharmacodynamic interaction
Pharmacodynamic interaction occurs when two drugs given together act at the same or similar receptor site and lead to a greater (additive or synergistic) effect or a decreased (antagonist) effect. ISDs, such as CsA, TAC, and SIR, block the critical events that lead to the activation of alloreactive T cells and the subsequent amplification of the signals involved in T cell proliferation. MMF inhibits the de novo synthesis of purines, which are necessary for the proliferation and function of T and B lymphocytes. PD-1 is a cell surface receptor that belongs to the immunoglobulin superfamily and is expressed on T cells and pro-B cells. 85 PD-1 binds two ligands, PD-L1, and PD-L2, and guards against autoimmunity through promoting apoptosis (programmed cell death) of antigen-specific T-cells in lymph nodes and reducing apoptosis in regulatory T cells. PD-1/PD-L1 inhibitors attack tumors by activating the immune system, 86 which potentially antagonize the immunosuppressive effect of ISDs. Though there are no published data on the potential risk, we do not suggest the application of PD-1/ PD-L1 inhibitors in KTRs.
Though acting at different receptor sites, ISDs and cytotoxic chemotherapy simultaneously enhance the systemic level of immunosuppression. Molecular targeted drugs have a very low risk of infection, and rarely increase the infection risk of chemotherapy. Nephrotoxicity, which is the potential ADR of CsA and TAC, 87 is also the potential ADR of cytotoxic chemotherapy and molecular targeting drugs. Hence, it is necessary to choose regimens with less risk of infection and nephrotoxicity.
Risk of infection
Infection is one of the major causes of death following renal transplant. The risk of infection in KTRs is determined by the synergy between two factors: the epidemiologic exposure of the individual and the ‘net state of immunosuppression’, which is a conceptual measure of all of the factors that contribute to the individual’s susceptibility (or resistance) to infection.88,89 Infections are most likely to occur between the 1st and 3rd month following transplant, since immune suppression is at its maximum due to induction therapy. 90 The risk of infection is relatively low due to stable and reduced levels of immunosuppression after 3 months of transplant. Cytotoxic chemotherapy adds to the risk of infection by suppressing the production of neutrophils, and by cytotoxic effects on the cells that line the alimentary tract.
The chemotherapy regimen is one of the primary determinants of the risk of neutropenia, and some regimens are more myelotoxic than others. Febrile neutropenia (FN) occurred in 17.6% (190/1078) of patients receiving FOLFIRI treatment, 13.4% (530/3957) of FOLFOX treatment, and 11.6% (165/1419) of 5-FU treatment. 91 CapeOx (0-1%) has been reported to be less myeloablative than FOLFOX and FOLFIRI. 92 As for monotherapy, the FN risk of capecitabine (0–1%) is lower than that of 5-FU CI (10.0–13.4%). 93 Molecular targeting drugs have very low risk of FN, and rarely increase the FN risk of chemotherapy. For metastatic CRC (mCRC) patients treated with FOLFOX and FOLFIRI regimens, the percentages of FN were 13.4% and 17.6%, respectively, while, combined with bevacizumab, the percentages were 11.8% and 13.3%, respectively. 91
Cytotoxic chemotherapy can adversely affect the developmental integrity of the gastrointestinal mucosa, which increases the risk of invasive infection due to colonizing bacteria or fungi that translocate across intestinal mucosal surfaces. Mucositis is the principal manifestation of mucosal toxicity related to chemotherapy. 94 Sonis and colleagues retrospectively analyzed mucositis in previous studies, and found the incidence of grade 3–4 oral mucositis in 5-FU/LV (14%, 95% CI 12–15%) is higher than that of FOLFOX (5%, 95% CI 4–7%), FOLFIRI (5%, 95% CI 3–8%), and FOLFOXIRI (6%, 95% CI 2–11%) regimens, while the incidence of gastrointestinal mucositis of FOLFOXIRI (38%, 95% CI 30–47%) is higher than that of FOLFIRI (25%, 95% CI 20–30%), 5-FU/LV (11%, 95% CI 10–12%), and FOLFOX (8%, 95% CI 6–10%) regimens. 94 As for oral versus intravenous fluoropyrimidines, there is no difference in the OR (0.64, 95% CI 0.25–1.62) for grade 3–4 mucositis between capecitabine based regimens, and 5-FU based regimens in adjuvant chemotherapy, while the pooled OR favored capecitabine-based regimens in palliative chemotherapy (OR 0.17, 95% CI 0.12–0.24). 95 Mucositis induced by molecular targeting drugs and immunotherapy drugs have not been reported in clinical trials.
Overall, no regimen for CRC is forbidden for KTRs when considering infection. Considering the immunosuppressive status of KTRs, it is wise to prevent or minimize the risk of infection. CapeOx and FOLFOX regimens have relatively low risk of FN and mucositis in the combined chemotherapy, while capecitabine has a relatively low risk in monotherapy. Adding molecular targeting drugs to chemotherapy does not significantly increase the risk of FN and mucositis. Special attention should be paid to infection during anticancer therapy.
Risk of nephrotoxicity
Nephrotoxicity is a potential ADR of anticancer pharmacotherapy and may result in a variety of functional abnormalities, including glomerular or tubular dysfunction, hypertension and disturbance of the renal endocrine function. In KTRs with CRC, the successful treatment of diseases might be limited by drug-related renal injury. The nephrotoxicity of these drugs are summarized in Table 4.
Nephrotoxicity of anti-cancer agents and ISD.
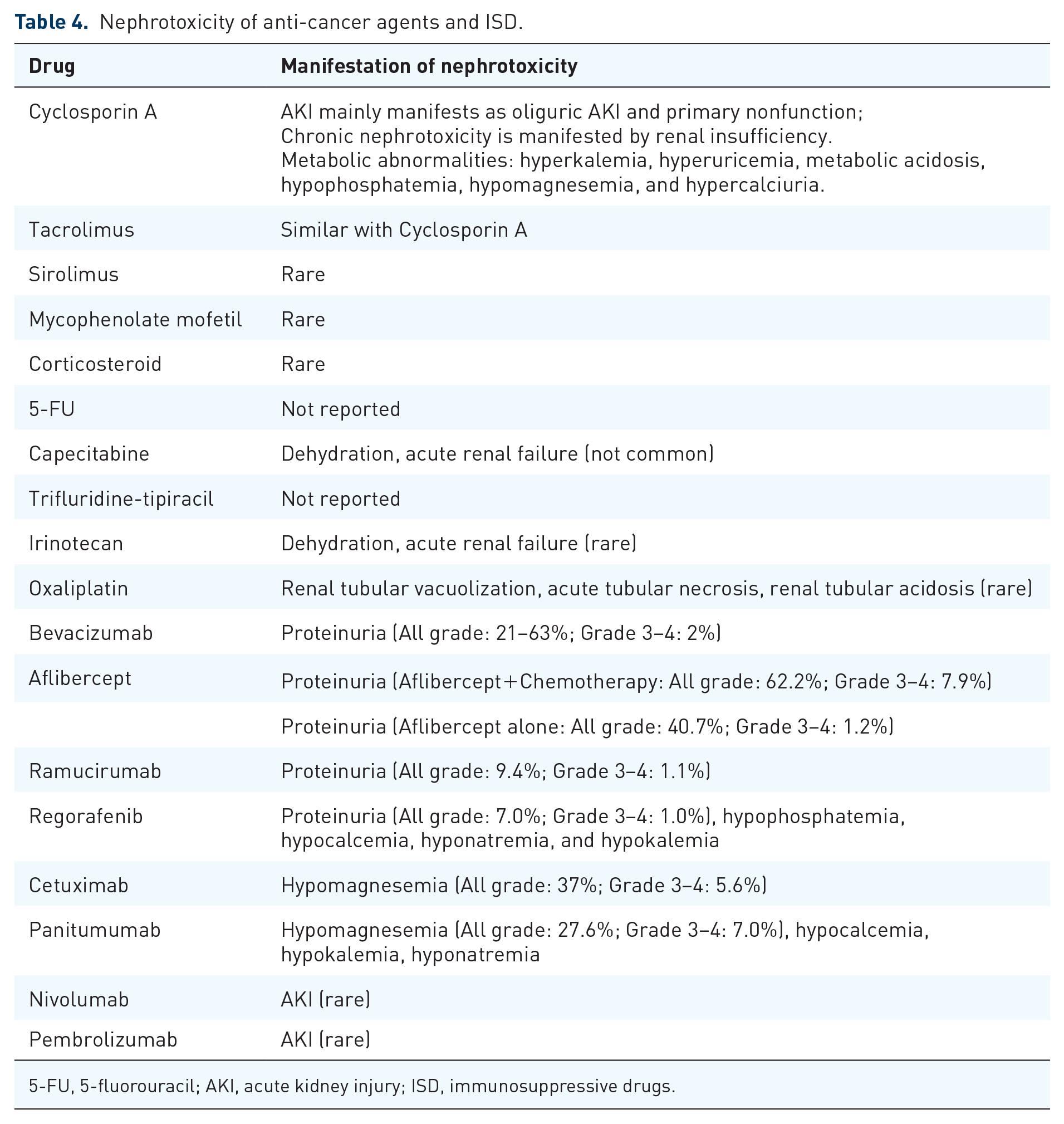
5-FU, 5-fluorouracil; AKI, acute kidney injury; ISD, immunosuppressive drugs.
Nephrotoxicity is rarely encountered with corticosteroids, SIR, and MMF, but is common with CsA and TAC. 87 Acute kidney injury (AKI) of CsA and TAC manifests mainly as oliguric AKI and primary nonfunction, 87 while chronic nephrotoxicity is manifested by renal insufficiency due to glomerular and vascular disease, abnormalities in tubular function, and an increase in blood pressure. 96 Trials comparing the risk of nephrotoxicity of CsA and TAC have shown contradictory results. Early researches found a similar incidence of early acute renal failure (ARF) and late hypertension, while late renal insufficiency was more prevalent with TAC.54,97 In contrast, some trials found that TAC was associated with less nephrotoxicity in liver transplants and similar nephrotoxicity in other transplants.42,98 Hence, it is generally considered that nephrotoxicity of TAC is generally similar to that of CsA. 47
CNIs, such as CsA or TAC, is necessary for the maintenance immunosuppressive therapy, so it is wise to minimize the nephrotoxicity of anticancer regimens. In patients with mild renal impairment (eGFR 50–70 ml/min), capecitabine treatment can induce diarrhea-related dehydration, and dehydration may cause acute renal failure. 99 Capecitabine-related renal failure rarely occurs in patients with normal renal function.
Vascular endothelial growth factor (VEGF) blockade is associated with proteinuria, which is rarely in the nephrotic range, and even more rarely associated with the nephrotic syndrome. 100 In patients treated with bevacizumab, the overall incidence of mild proteinuria ranges from 21% up to 63%, and incidence of grade 3 or 4 proteinuria is about 2%. 101 Aflibercept plus chemotherapy led to 62.2% of all-grade proteinuria and 7.9% of grade 3 or 4 proteinuria, while aflibercept alone led to 40.7% of all-grade proteinuria and 1.2 % of grade 3 or 4 proteinuria in a phase III trial. 101 Among patients treated with ramucirumab, the risk of proteinuria may be lower. In a meta-analysis of six placebo-controlled randomized trials, the incidence of all-grade proteinuria for ramucirumab versus placebo was 9.4% versus 3.1%, while the risk of severe (grade 3 or 4) proteinuria was 1.1% versus 0.04%. 102 Previous studies have shown contradictory results on the proteinuria risk of regorafenib. The United States package insert lists the rate of all grade proteinuria for single agent regorafenib to be 51–84% compared with 37–61% in the placebo treatment arm. In a phase III trial involving 500 patients receiving regorafenib, the incidence of all-grade proteinuria was 7.0% and grade 3 or 4 proteinuria was 1.0%. 103
Bevacizumab is recommended as a first-line antiangiogenic drug in National Comprehensive Cancer Network (NCCN) guidelines, but ramucirumab seems to have the lowest risk of nephrotoxicity. It is hard to decide the best antiangiogenic drugs for CRC in KTRs. Anti-VEGF associated proteinuria is usually an asymptomatic event detected only through laboratory analysis. Hence, all antiangiogenic drugs are applicable for CRC in KTRs, and bevacizumab might be the first-line choice.
Cetuximab and panitumumab are both associated with the progressive development of hypomagnesemia due to renal magnesium wasting.95,96 Nephrotoxicity of cetuximab has also been manifested as hypocalcemia, hypokalemia, and hyponatremia.104–106 In a meta-analysis of 19 clinical reports, cetuximab-based treatment led to 37% of all-grade hypomagnesemia and 5.6% of grade 3 or 4 hypomagnesemia. 104 Panitumumab led to 27.6% of all-grade hypomagnesemia and 7.0% of grade 3 or 4 hypomagnesemia in patients with chemotherapy-refractory mCRC. 95 Hypomagnesemia resolves after treatment is discontinued. Hence, cetuximab and panitumumab are equally applicable for CRC in KTRs.
Clinically significant renal toxicity induced by chemotherapy regimens is rare in CRC patients. Adding molecular targeted drugs to chemotherapy increase the risk of nephrotoxicity, but is feasible. We suggest measuring urine volume, urine protein excretion, and serum creatinine after every cycle of anticancer therapy or every 1 month. Glomerular filtration rate (GFR) should be estimated whenever serum creatinine is measured. We also suggest kidney allograft ultrasound examination as part of the assessment of kidney allograft dysfunction.
Other considerations in antitumor pharmacotherapy
Fluoropyrimidine-related cardiotoxicity
Fluoropyrimidine-related cardiotoxicity is an infrequent but potentially lethal ADR. The most common clinical manifestation is chest pain, which can be either nonspecific or anginal and is usually associated with electrocardiographic (ECG) changes. 107 Symptoms occur with or without elevated serum biomarkers of cardiac injury. Chest pain tends to occur most often in the first cycle of administration, but can be delayed. The incidence of cardiotoxicity in capecitabine (3–9%)108,109 is within the range of that reported with infusional 5-FU (1–19%).107,110,111 The incidence in CapeOx regimens (12% in one report) may be higher than with capecitabine alone. 108 Cardiotoxicity appears to be completely reversible after termination of FU treatment. 107 The preferred strategy in most cases is to switch to a nonfluoropyrimidine-containing chemotherapy regimen or a different treatment modality.
Hypertension of antiangiogenesis drugs
All commercially available angiogenesis inhibitors have been implicated in the development of hypertension. Previous studies have shown similar incidence of all-grade hypertension (24% versus 21.3%) and severe hypertension (8.0% versus 9.0%) between bevacizumab and ramucirumab.102,104 The incidence of severe hypertension may be higher with aflibercept (aflibercept + chemotherapy 19.1% versus chemotherapy 1.5%) than with other VEGF-targeted therapies, but no direct comparisons of aflibercept with other VEGF inhibitors exist. 112 All patients receiving angiogenesis inhibitor should have their blood pressure actively monitored during treatment, with more frequent measurement in the first few weeks of therapy. Manage blood pressure during therapy, with an ideal goal, where possible, of keeping blood pressure less than 130/80mmHg for most patients, and lower in those with specific pre-existing cardiovascular risk factors, such as diabetes or chronic kidney disease.
Skin toxicity
Given the fact that EGFR is expressed in skin and adnexal structures, EGFR inhibitors are associated with skin toxicity, mainly acneiform eruption and xerosis, but also paronychia, abnormal scalp, facial hair, and eyelash growth, maculopapular rash, mucositis, and postinflammatory hyperpigmentation.113,114 PRIDE syndrome comprises the most frequent reactions associated with anti-EGFR reactions (papules, pustules, paronychia, hair growth disorders, pruritus, and skin and mucosal xerosis). 114 In a recent meta-analysis involving 38 studies, any grade skin toxicities were identical in cetuximab and panitumumab (83.6% versus 84.4%; p = 0.99). However, cetuximab was associated with fewer G3-4 skin toxicities (15.7 versus 23.2%; OR = 0.62, 95%CI 0.53–0.62; p < 0.001). 115 Conversely, cetuximab was associated with slightly more frequent G3-4 acne-like rash (16 versus 13%; OR = 1.24, 95% CI 1.04–1.48; p = 0.04) and paronychia (23 versus 18%; OR 1.36, 95% CI 1.1–1.7) but fewer cases of all-grade skin fissures (8.5 versus 12.8%; OR = 0.64, 95% CI 0.44–0.93; p = 0.02) and all-grade pruritus (17.4 versus 32%; OR = 0.45, 95% CI 0.35–0.58; p < 0.001) than panitumumab. 115 Incidence of skin toxicity of EGFR inhibitor in renal transplant recipients has not been reported. Because of the common use of corticosteroids, the incidence and severity of EGFR-inhibitor-induced acneiform eruption in kidney recipients might be lower in kidney recipients than in nontransplant patients.
The initial pustules are sterile, but secondary infection of the affected skin may occur. Eilers and colleagues reported that 38% (84 of 221) of patients treated with EGFR inhibitor developed infections with bacteria, dermatophytes, or viruses. 116 Staphylococcus aureus was the most common pathogen, found in approximately 60% of infected lesions. Secondary infection may result in sudden worsening of cutaneous rashes and an alteration in clinical status. 114 Hence, preventive measures are needed, such as adequate hydration of dry areas, decrease in sun exposure, avoiding prolonged skin contact with water, irritants, and solvents. Topical agents can reduce reaction severity. Moderate reactions may be treated with oral antibiotics, including tetracycline, doxycycline. and minocycline besides topical agents. Tetracycline is not recommended for kidney recipients due to its nephrotoxicity. Doxycycline and minocycline can be used in these patients, but the dose of minocycline should be reduced in patients with renal injury.
Paronychia and nail lesions occur in 25% of patients treated with panitumumab and 16% of those treated with cetuximab. 113 Avoiding friction and pressure on the nailfold, and application of topical steroids, are commonly used to reduce reaction severity. Paronychia is not considered an infectious process, but secondary infection with S. aureus or coagulase-negative, Gram-positive bacteria (nosocomial colonization) can occur. 117 In this condition, topical antimicrobial agents (mupirocin or nystatin ointment ) and oral doxycycline might be used.
Neurotoxicity of oxaliplatin
The major dose-limiting toxicity with oxaliplatin is neurotoxicity. There are two distinct syndromes: acute neurotoxicity and cumulative sensory neuropathy. Acute neurotoxicity develops within 24–72 h after each dose and recurs with each dose. 118 Symptoms consist of striking paresthesias and dysesthesias of the hands, feet, and perioral region, jaw tightness, and unusual pharyngo-laryngo-dysesthesias.119,120 Patients need to be warned not to drink cold fluids in the days around their oxaliplatin infusions. Lengthening the infusion duration from 2 h to 6 h can prevent recurrence of the pseudolaryngo spasm.
Late-onset neuropathy with oxaliplatin is cumulative, dose-dependent, and typically consists of a sensory, symmetric distal axonal neuropathy without motor involvement. Incidence and severity are related predominantly to cumulative dose. For example, the incidence of grade 3 neuropathy with cumulative doses of 850 mg/m2 is 10–15%, and rises thereafter. As the clinical situation permits, if significant neuropathy develops during treatment, we suggest discontinuing oxaliplatin and switching to a non-oxaliplatin chemotherapy to permit as much recovery as possible before reintroducing oxaliplatin.
Experience of pharmacotherapy on post-transplantation CRC
Pharmacotherapy is of particular important for the treatment of CRC. The median overall survival (OS) is about 6 months in untreated mCRC. The OS has been extended up to 12–20 months by treatment with combined chemotherapy such as FOLFOX, FOLFIRI, and CapeOx. The OS has been further improved to 24–41 months after adding target agents. 34 A few cases have explored the pharmacotherapy of CRC in KTRs (Table 5).1, 121–124
Parameters of patients with CRC after renal transplantation.

5-FU, 5-fluorouracil; ADR, adverse drug reaction; Aza, azathioprine; CRC, colorectal cancer; CSA, cyclosporin A; EVE, Everolimus; ISD, immunosuppressive drugs; MMF, mycophenolate mofetil; P, prednisone; PD, programmed cell death; TAC, Tacrolimus.
Modified chemotherapy, such as single agent therapy, was attempted to improve the survival and compliance of CRC in KTRs. In a study by Liu and colleagues, three cases of advanced rectal cancer were treated with capecitabine adjuvant chemotherapy after surgery. 1 Gu and colleagues reported a case of a 57-year-old female with advanced rectal cancer who received three cycles of oxaliplatin chemotherapy. 122 Trivedi and colleagues also reported the application of continuous 5-FU infusion in a colon cancer patient. Chemotherapy was well tolerated in all these patients. 124
The application of combination therapy for post-transplantation CRC has been explored. In our previous study, an advanced colon cancer was treated with three cycles of FOLFOX chemotherapy. Plasma concentration of ISDs (CsA and SIR) and renal function were not affected by chemotherapy. No serious ADR was observed. 121
Targeted therapy also has been reported. Müsri and colleagues reported a case of post-transplantation mCRC treated with bevacizumab + FOLFIRI. The dose of bevacizumab was 5 mg/kg/day for 14 days. Proteinuria (2.5 g/day) existed before the start of the treatment, and increased to 4 g/day at the end of the fifth course. 123 It is wise to exclude patients with proteinuria before the start of antiangiogenic therapy.
Potential management strategies
Immunosuppressive management in KTRs with CRC
In KTRs with CRC, immunosuppressive therapy and antitumor therapies are both important, and constrict each other. Disturbance of immunosuppressive level, either under-immunosuppression or over-immunosuppression, can lead to other serious complications, such as graft rejection, infection, and renal dysfunction. Those are also contraindications for antitumor therapy. Preservation of graft function is of primary importance, and informs the basis of antitumor therapy. Hence our suggestions of ISDs for KTRs with CRC are similar to KDIGO clinical practice guidelines, which recommend TAC as the first-line CNI, and MMF as the first-line antiproliferative agent. CsA is recommended as the alternative CNI, and SIR as the alternative antiproliferative agent.
Although lacking trial-based evidence, judicious reduction in immunosuppression with regular monitoring of disease progression and graft function may be warranted, particularly among those with high-grade and advanced malignancy. We suggest KTRs with CRC reach the trough concentration recommended by KDIGO guideline for KTRs after 1 year of renal transplant, which is that the trough concentration of TAC should be more than 5–10 ng/ml. We also recommend intensive therapeutic drug monitoring of ISD such as testing of trough concentration before and after anticancer therapy.
Antitumor management in KTRs with CRC
For patients with indication of adjuvant chemotherapy, FOLFOX or CapeOx is preferred for patients who can tolerate combined chemotherapy, and 5-FU/LV or capecitabine is recommended for those patients who cannot tolerate combined chemotherapy. These regimens are also recommended as first-line palliative chemotherapy, and can be combined with molecular targeted drugs (anti-angiogenesis drugs and anti-EGFR MoAbs). FOLFIRI only can be used in oxaliplatin-refractory patients. In subsequent therapy, aflibercept, trifluridine-tipiracil, and tegafur-uracil are suggested for these patients.
Pharmaceutical care in KTRs with CRC
Regardless of the antitumor regimen, trough concentration of ISDs, renal function, blood routine, and cardiac function should be intensively monitored. DPD and TYMS genotype should be detected in order to make an early decision regarding fluoropyrimidines dose reduction. Attention should be paid to neurotoxicity if an oxaliplatin-based regimen (CapeOx or FOLFOX) is chosen. Blood pressure and urine protein excretion should be intensively monitored if an antiangiogenic drug is added. Attention should be paid to hypomagnesemia and skin toxicity if anti-EGFR MoAb is added.
Irinotecan has the risk of severe diarrhea, which substantially increased the exposure of TAC. Irinotecan also has the potential of competing for UGT1A, which might lead to the accumulation of MMF and irinotecan. Hence irinotecan-based regimens (FOLFIRI and FOLFOXIRI) are not preferred, but can be used in oxaliplatin-refractory patients. UGT1A1 genotype should be determined before the initiation of irinotecan-based regimens, and the starting dose of irinotecan should be lowered in patients known to be homozygous for UGT1A1*28. Cetuximab or panitumumab combined with FOLFIRI are not recommended for the synergistic effect of diarrhea.
Nivolumab or pembrolizumab should be avoided due to their immune enhancement effect. Regorafenib is not recommended because of its inhibiting effect on UGT1A and potential competition for CYP3A4. Attention should be paid to the interaction between ISDs and PPIs. All PPIs reduce absorption of MMF, and omeprazole might inhibit CYP3A4-mediated metabolism of TAC .
Conclusion
Antitumor pharmacotherapy of CRC in KTRs is a dilemma. Impediments such as drug interactions and potential ADR really exist. But these should not be contraindications for KTRs to adopt anticancer therapy. Toxicity can be minimized by selecting regimens and managing ADR, and a few cases have also proved the feasibility of pharmacotherapy in these patients. We also provide potential antitumor regimens for the adjuvant, palliative, and subsequent therapy of CRC in KTRs. Future clinical trials or case reports are needed to confirm the relative benefit and risk of antitumor pharmacotherapy in KTRs.
Footnotes
Acknowledgements
Authors Yuanyuan Fu and Chengheng Liao contributed equally.
Funding
The author(s) received no financial support for the research, authorship, and publication of this article.
Conflict of interest statement
The authors declare that there is no conflict of interest.