
Abstract
Patent expirations for several biological products have prompted the development of alternative versions, termed ‘biosimilars’, which have comparable quality, safety and efficacy to a licensed biological medicine (also referred to as the ‘reference’ medicine). The first biosimilars developed in oncology were the supportive-care agents filgrastim and epoetin. Binocrit® (HX575) is a biosimilar version of epoetin alfa, indicated in the oncology setting for the treatment of chemotherapy-induced anemia (CIA). The process for development and approval of Binocrit® as a biosimilar included extensive analytical characterization and comparison with the reference epoetin alfa. This was followed by a clinical development program comprising phase I pharmacokinetic/pharmacodynamic studies to show bioequivalence to the reference medicine and a confirmatory phase III study to confirm therapeutic effectiveness in CIA. Since its approval, Binocrit® has been extensively used and studied in real-world clinical practice. The accumulated data confirm that Binocrit® is an effective and well-tolerated option for the treatment of CIA in patients with cancer.
Introduction
Biological medicines are important therapeutic options and include supportive-care agents and drugs active in many therapeutic areas such as cancer and immune-mediated inflammatory diseases. Patent expirations for several biological products have prompted the development of alternative versions, termed ‘biosimilars’, which have comparable quality characteristics, biological activity, safety and efficacy to a licensed biological medicine (also referred to as the ‘reference’ medicine) and are associated with lower development costs. 1 Biosimilar medicines may offer an opportunity to positively impact on the financial sustainability of healthcare systems. 2 By some estimates, the introduction of biosimilars may result in cumulative savings [across the United States (US), France, Germany, Italy, Spain and the United Kingdom (UK)] of up to 100 billion Euros by 2020. 3
This review will outline the processes associated with biosimilar development and approval using the example of the first biosimilar epoetin approved in Europe, Binocrit®. Since it was approved in 2007, there is now a decade of clinical experience with this biosimilar medicine.
Biosimilar development and approval
The development of high-quality biosimilars is a systematic and robust process involving several steps (Figure 1), and approval takes account of the totality of evidence. The first stage consists of thorough molecular characterization of the reference medicine. 4 This involves defining the critical features (or quality attributes) that determine the clinical properties [efficacy and safety, pharmacokinetics (PK) and pharmacodynamics (PD), immunogenicity] of the reference medicine and obtaining information about the extent to which these attributes vary and might change over time. These data are the basis to define the boundaries, or ‘goal posts’, within which the attributes of the biosimilar in development must fall. 4 As part of this exercise, a comprehensive panel of analytical methods is utilized to ensure that the biosimilar has the same structural and functional properties as the reference medicine.4,5

Overview of the biosimilar development process. 4
The goal of the clinical program is not to demonstrate new safety and efficacy attributes, as this has already been established for the reference medicine, but to convincingly confirm the absence of clinically meaningful differences as compared with the reference medicine. Clinical development of biosimilars usually includes phase I PK/PD studies to show bioequivalence and a phase III confirmatory study in a selected sensitive indication to demonstrate that there are no meaningful clinical differences compared with the reference medicine. Regulatory authorities will approve a proposed biosimilar only if similarity to the reference medicine is established in terms of quality characteristics, biological activity, and safety and efficacy based on the totality of evidence.6,7 Following the approval of a biosimilar medicine, standard pharmacovigilance is required (as with any new medicines), especially given that immunogenicity is important to monitor for all biologics. 8
The European Medicines Agency (EMA) was the first regulatory body to develop a specific regulatory pathway for the approval of biosimilars when it published ‘Guidelines on similar biological medicinal products’ in 2005. 6 Numerous additional updates and guidelines have since been published by the EMA 9 and other regulatory authorities including, more recently, the US Food and Drug Administration in 2015. 7
To date, the EMA has approved two biosimilar epoetins under several brand names. A biosimilar version of Eprex®/Erypo® (Janssen-Cilag, High Wycombe, UK), which has the same international nonproprietary name (INN; epoetin alfa) is marketed as Binocrit® (Sandoz GmbH, Kundl, Austria), Epoetin alfa HEXAL® (Hexal, Holzkirchen, Germany) and Abseamed® (Medice Arzneimittel, Iserlohn, Germany). Another biosimilar version of Eprex®/Erypo® is also available, with an INN of epoetin zeta; this is marketed as Retacrit® (Hospira UK Limited, Maidenhead, UK) and Silapo® (STADA, Bad Vilbel, Germany).
Development and approval of Binocrit® in Europe
Eprex®/Erypo® (epoetin alfa), a recombinant human erythropoietin, was licensed in 1989 and indications now include anemia associated with renal failure, chemotherapy-induced anemia (CIA) and moderate anemia prior to major elective orthopedic surgery where there is a high risk for perioperative transfusions. 10 In 2007, Binocrit® became the first biosimilar epoetin alfa to be approved in Europe, with Eprex®/Erypo® used as the reference medicine.
Analytical characterization
Extensive characterization (physicochemical and biological) is a key aspect of the biosimilar approval process. For Binocrit®, this characterization included an array of state-of-the-art tests for protein structure, presence of aggregates, isoform content, receptor binding and biological activity. 11 This range of analytical methods demonstrated the similarity of Binocrit® and the reference epoetin alfa in terms of primary protein structure, higher-order protein structure (Figure 2), isoform pattern, post-translational modifications, receptor binding and biological activity.

Comparison of (a) far- and near-UV CD spectra and (b) peptide mapping profiles for Binocrit® and reference epoetin alfa. 11 (Reproduced with permission from European Association of Hospital Pharmacists.)
Biosimilar manufacturers are also able to take advantage of technological improvements, and they use the latest available systems to produce and purify biosimilar proteins. Manufacturers of reference medicines have also adapted their production methods but have older technologies as a basis, due in part to the financial and regulatory impact of making changes to their methods.12,13 The technical quality of biosimilar epoetins has thus been shown to exceed that of the reference medicines in some attributes; for example, Binocrit® was shown to have lower levels of certain impurities compared with the reference medicine. 14
Clinical development: phase I PK/PD studies
The clinical development program for Binocrit® included two open-label, randomized, parallel-group phase I studies which evaluated the PK and PD properties of Binocrit® and those of the reference medicine following intravenous (IV) or subcutaneous administration in healthy volunteers (n = 80 per study).15,16 In these studies, Binocrit® and the reference epoetin alfa were found to be bioequivalent in relation to their PK profiles. Further, Binocrit® met the PD primary endpoint of predefined biosimilarity criteria with respect to the area under the total effect curves (AUEC) during 12 dosage intervals in 4 weeks for hemoglobin (Hb). Multiple doses of study medication were well tolerated, with similar safety profiles across the treatment groups. No anti-epoetin antibodies were detected with either treatment.15,16
Clinical development: phase III confirmatory studies
A study in patients (n = 114) with solid tumors and CIA demonstrated the efficacy of Binocrit® in this setting. 17 Patients were randomized to treatment with Binocrit® (n = 74) or the reference medicine (Eprex®/Erypo®; n = 40) with data from the control arm serving as an internal control during secondary analyses. The time course of Hb levels is shown in Figure 3. The majority of patients in the Binocrit® group (62%) had a Hb increase of at least 2 g/dl from mean baseline/screening value to weeks 5–12 without red blood cell (RBC) transfusions in the preceding 4 weeks (Table 1). The confidence interval (CI) was entirely above the predefined threshold of 30%, confirming that the primary endpoint was met. Similar results were noted with both treatments for secondary efficacy measures, including transfusion requirements (32% of patients in the Binocrit® group and 38% of those in the control group). The safety profile of the two treatments was also similar; the incidence of serious adverse events (AEs) was comparable in both groups, and the incidence of drug-related AEs was lower in the Binocrit® group (19%) compared with the control group (32%). No anti-epoetin antibodies were detected.
Main Hb outcomes in studies reporting data on Binocrit® for the treatment of chemotherapy-induced anemia.

Hb increase of at least 2 g/dl from mean baseline/screening value to weeks 5–12 without red blood cell transfusions in the preceding 4 weeks.
Increase of at least 1 g/dl in 4 weeks or Hb in the range 10–12 g/dl during the study.
Minor response defined as Hb increase of 1–2 g/dl, major response as Hb increase of >2 g/dl.
Hb, hemoglobin.
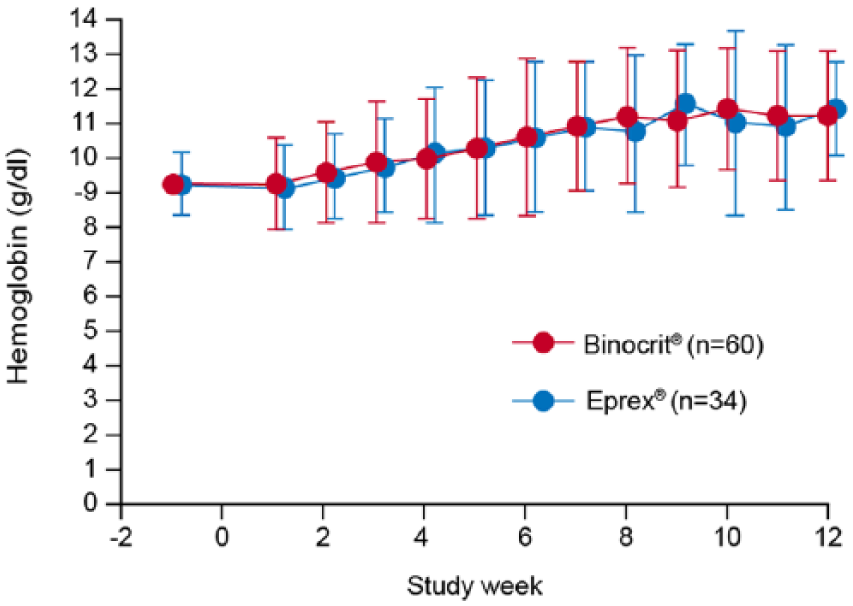
Time course of hemoglobin levels. 15 (Reproduced with permission from Karger.)
Another controlled study established the therapeutic equivalence and long-term efficacy and safety of Binocrit® for the treatment of anemia in hemodialysis patients with chronic kidney disease. 25 Therapeutic equivalence of Binocrit® and the reference epoetin alfa was statistically confirmed: mean changes in Hb levels were 0.15 ± 0.09 g/dl in the Binocrit® and 0.06 ± 0.12 g/dl in the reference epoetin alfa, with a difference between groups of 0.08 g/dl (95% CI −0.17 to 0.34). The long-term (56-week) safety profile of Binocrit® was also similar to that of the reference medicine and there was no evidence of neutralizing antibodies or immunogenicity in either treatment group.
Further clinical experience with Binocrit®
In addition to these registration studies, Binocrit® has been extensively studied in real-world clinical practice. Table 1 shows the published clinical studies (confirmatory phase III and post-approval) of Binocrit® for the treatment of CIA. The major tumor types included in these studies are summarized in Figure 4.
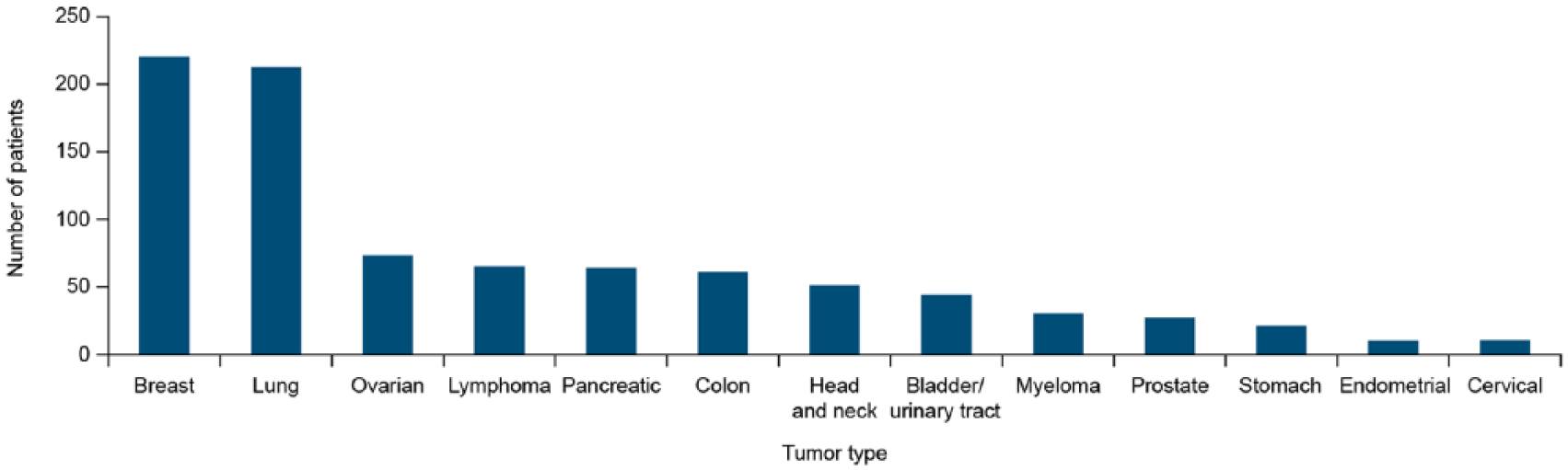
Most frequent cancers included in studies of Binocrit® for the treatment of chemotherapy-induced anemia.
A retrospective clinical audit has assessed the effectiveness of Binocrit® for the treatment of CIA in patients (n = 152) with solid tumors. 18 Data were collected from patients at five centers in Europe (one each in France, Italy, The Netherlands, Romania and Spain). In the overall population, 74% of patients achieved a Hb response, defined as an increase of at least 1 g/dl in 4 weeks or Hb in the range 10–12 g/dl during the study. Among evaluable patients (n = 113, those with a baseline Hb ⩾8.5 g/dl who received epoetin treatment for at least 6 weeks), the response rate was 79% (Table 1). Response rates were similar among evaluable patients who received an initial Binocrit® dose of 30,000 or 40,000 IU/week (81% versus 78%, p = ns) and significantly greater in patients who also received IV iron (93% versus 77% in those who did not receive IV iron, p < 0.05). No serious unexpected drug-related AEs were reported.
Single-center experiences with Binocrit® from Germany and Spain have also been published. 19 For the Spanish center, a total of 274 patients with various solid tumors were included in the analysis. Patients were treated with Binocrit® 40,000 IU (n = 116) or 30,000 IU (n = 14) once weekly, darbepoetin alfa 500 µg once every 3 weeks (n = 99) or darbepoetin alfa 150 µg once weekly (n = 45). Mean overall Hb prior to treatment was 9.3 g/dl, which increased to 10.8 g/dl by the end of the study. There were no significant differences between the treatments in terms of Hb level at the start of treatment, Hb level achieved at the end of the treatment period or the highest Hb level achieved. The proportion of patients overall who required a transfusion was low (13%, 38 patients) and generally similar across the different treatments, and no serious drug-related AEs were reported in any group.
For the German center, a retrospective matched-cohort analysis was conducted on 145 patients with solid tumors (122 of whom had breast cancer). 19 Patients were treated with Binocrit® 40,000 IU once weekly (n = 95) or darbepoetin alfa 500 µg once every 3 weeks (n = 50). There were no significant differences between the treatment groups in Hb level at the start or end of treatment. In both groups, the median time to achieve a Hb increase >1 g/dl and >2 g/dl was 2 and 4 weeks, respectively. Overall, four patients in each group required a RBC transfusion during the period of erythropoiesis-stimulating agent treatment. No deaths, thromboembolic events or other serious adverse drug reactions were observed in either treatment group.
An analysis was also conducted of pooled data from the two centers for Hb outcomes and transfusion requirements (Table 1). The mean increase in Hb was 1.98 g/dl with Binocrit® and 1.82 g/dl with darbepoetin alfa (p = 0.5). A total of 18 patients (8%) in the Binocrit® group and 28 patients (14%) in the darbepoetin group required a blood transfusion (p = 0.039). 19
OncoBOS is an ongoing, prospective, observational study of the use of Binocrit® for the treatment of CIA being conducted in France. Versions of the study have also been taken up in Germany, Italy, Romania and Austria. An evaluation of 1298 patients with solid tumors in the French study has been reported. 20 Mean Hb at baseline was 9.7 g/dl, increasing to 10.6 g/dl at week 3–4 and 11.1 g/dl at week 12 (p < 0.001 versus baseline; Table 1). The required dose of Binocrit® remained stable over the treatment period and 81% of patients in this evaluation were able to continue their chemotherapy without delays or dose reductions. 20 Data have also been reported from the Italian study named ANEMONE. 21 A total of 245 patients were enrolled, with 215 evaluable for statistical analysis. In the first 4 weeks, 49.3% of patients showed an increase in Hb of ⩾1 g/dl (45.5% in patients with solid tumors and 52.1% in patients with hematological malignancies). In the first 12 weeks, 51.6% of patients showed an increase in Hb of ⩾2 g/dl (48.4% solid tumors, 54.2% hematological diseases). Treatment with Binocrit® was well tolerated. 21
A number of other studies have also assessed the use of Binocrit® to treat CIA in onco-hematological patients. In one study, Binocrit® was used alongside a systematic approach to iron supplementation based on iron status markers (including the novel marker zinc protoporphyrin) in patients (n = 19) with hematological malignancies. 22 Of the 19 patients, 18 had a response to Binocrit® (Table 1); 12 of these had a major response (Hb increase >2 g/dl) and 6 had a minor response (Hb increase 1–2 g/dl). Another study has reported on the efficacy and safety of Binocrit® for the treatment of CIA in patients (n = 65) with chronic lymphoid neoplasms. 23 Mean (standard deviation) Hb level at the initiation of treatment was 9.3 (0.5) g/dl. Mean Hb levels increased to 10.7 (1.4) and 10.6 (1.5) g/dl (patients on first-line chemotherapy), and 11.4 (1.6) and 9.7 (1.3) g/dl (those on salvage chemotherapy), at weeks 4 and 8, respectively. Treatment with Binocrit® was well tolerated and allowed patients with non-Hodgkin lymphoma or chronic lymphoproliferative disorders to continue their course of chemotherapy by effectively increasing and maintaining Hb levels. 23 Binocrit® treatment (for a minimum of 12 weeks) has also been evaluated in a small study of elderly patients (age ≥65 years; n = 31) with multiple myeloma. 24 Transfusion requirements were significantly decreased regardless of patients’ transfusion history and Hb levels were significantly increased (median Hb 8.20 g/dl and 9.40 g/dl before and after treatment; p < 0.001). Treatment with Binocrit® also provided improvements in social-relational functioning and cognitive wellbeing. 24
A preliminary study has assessed the effectiveness of Binocrit® for the treatment of anemia in low-/intermediate-1 risk myelodysplastic syndromes. 26 A total of 24 consecutive patients aged > 65 years were treated with Binocrit® at 40,000 IU once weekly for 12 weeks and were followed for at least 3 months. Responsive patients continued with 40,000 IU once weekly for a further 12 weeks. Overall, 16 patients (66.67%) achieved an erythroid response (either an increase of Hb ⩾1.5 g/dl lasting for ⩾8 weeks or a reduction of >4 units of packed red cell transfusions in a period of 8 weeks for patients with transfusion-dependent anemia); 15 patients (62.5%) became transfusion-independent and remained free from transfusion requirement for at least 3 months, while 2 patients had reduction in transfusion requirement of at least four RBC transfusions/8 weeks compared with the pretreatment transfusion requirement. 26
Other published data include those from a prospective, randomized study in healthy volunteers that compared Binocrit® with a US-marketed epoetin alfa (Epogen®) and with the European reference medicine (Eprex®/Erypo®). 27 All products were well tolerated and had a similar safety profile, with no reports of anti-erythropoietin antibodies. Similarly, a review of clinical safety data for biosimilar epoetins found no difference in safety profiles between biosimilar and reference medicines, nor any between the different biosimilar epoetin products. 28
Summary
Biosimilars, including biosimilar epoetin alfas, have now been available in Europe for approximately 10 years. These agents are approved by the EMA only if they are shown in extensive analytical and clinical testing to have comparable quality characteristics, biological activity, safety and efficacy to the reference medicine. Binocrit®/HX575 is a biosimilar epoetin alfa used in the oncology setting for the treatment of CIA. Since its approval, Binocrit® has been extensively studied in real-world clinical practice. The data available are reassuring that Binocrit® is an effective and well-tolerated option for the treatment of CIA in patients with cancer. Furthermore, routine ongoing pharmacovigilance has not raised any unexpected or additional safety signals (beyond those known for the reference medicine) associated with Binocrit® following 10 years of clinical usage in Europe. As of April 2016, Binocrit® has generated more than 400,000 patient years of experience worldwide. 28 Moreover, ongoing studies will continue to extend the evidence base in clinical settings, such as CIA. The availability of biosimilar medicines offers affordable, high-quality, effective alternative treatments and may help contain healthcare budgets while improving treatment access for patients.
Footnotes
Funding
This work was supported by Sandoz/Hexal AG.
Editorial support was provided by Tony Reardon, Spirit Medical Communications Ltd., supported by Hexal AG.
Conflict of interest statement
MA and PG have acted as advisors to Sandoz. AK, NH and AS are current or previous employees of Sandoz/Hexal AG.