
Abstract
Background and objectives:
This phase III study (RI-01-006; FLINTER) was conducted to demonstrate equivalent efficacy of DRL_RI to EU-approved rituximab (MabThera®) in patients with previously untreated Stage II–IV, CD20-positive, low-tumor-burden follicular lymphoma (LTB-FL). This study also evaluated safety, immunogenicity, rituximab concentrations, and pharmacodynamics (PD) of DRL_RI compared with MabThera.
Design and methods:
Previously untreated, stage II–IV, CD20-positive LTB-FL patients (N = 317) were randomized (1:1) to receive DRL_RI (n = 162) or MabThera (n = 155) as intravenous infusions of 375 mg/m² weekly for 4 weeks (induction period), and thereafter every 8 weeks from Week 12 to Week 36 (maintenance treatment), and followed up till Week 52. The primary end point was best overall response rate (BORR) up to Week 28 based on blinded independent central review. Efficacy equivalence was demonstrated if the two-sided 90% confidence interval (CI) for BORR difference was within the prespecified equivalence margin (±17%). Secondary end points included objective and complete responses, duration of response, progression-free survival, overall survival, safety, immunogenicity, mean serum concentrations, and PD.
Results:
The BORR up to Week 28 was 80.2% versus 79.4% for DRL_RI versus MabThera group; with a difference of 0.89% (90% CI: −6.67 to 8.48; 95% CI: −8.05 to 9.93 within the prespecified margin). Both treatment groups were comparable for all secondary efficacy end points. Treatment-emergent adverse events were reported in 68.6% of patients; safety, immunogenicity, and mean serum concentrations were similar between groups. Peripheral B-cell counts declined below quantifiable limits in most patients, with a median time to B-cell depletion of 6.9 versus 7.0 days for DRL_RI versus MabThera.
Conclusion:
The study demonstrated efficacy equivalence of DRL_RI to MabThera; with comparable safety, immunogenicity, serum concentrations, and PD between groups.
Trial registration:
This trial was registered at ClinicalTrials.gov identifier: NCT03976102 and EudraCT (2018-004223-36).
Keywords
Introduction
Non-Hodgkin lymphoma (NHL), the most common hematological malignancy in adults, includes a heterogeneous group of B-cell lymphomas that account for almost 85% of all NHL diagnoses.1,2 The risk of mortality from NHL depends on whether it is an indolent or aggressive subtype; the indolent form being slowly progressing yet incurable. Follicular lymphoma (FL) is the most common subtype of indolent B-cell NHL diagnosed in the Western hemisphere, comprising 70% of indolent and 22% of all NHLs.3,4 Rituximab (Rituxan® and MabThera®) [Roche Diagnostics GmbH, Germany], the chimeric anti-CD20 monoclonal antibody (mAb), has been widely studied in patients with indolent lymphoma following its approval over two decades now for use in various B-cell lymphomas.5,6 Rituximab monotherapy has been considered for first-line treatment in patients with low-tumor-burden follicular lymphoma (LTB-FL) by the European Society for Medical Oncology clinical practice guidelines and the U.S. National Comprehensive Cancer Network guidelines.7,8
Dr. Reddy’s Laboratories Ltd (DRL) has developed a biosimilar of rituximab, hereafter referred to as DRL_RI. While the definitions for biosimilarity vary slightly between the European Medicines Agency (EMA) and the U.S. Food and Drug Administration (U.S. FDA), fundamentally, biosimilars are defined as biologic medicines highly similar to the licensed originator/reference product, comparable in terms of purity, quality, safety, and efficacy with no clinically meaningful differences against the original biologic.9,10 DRL_RI is a chimeric human/murine IgG1 kappa mAb consisting of murine light and heavy chain variable regions and human constant region sequences. As part of the stepwise development plan for biosimilars, an extensive comparison of structural, physicochemical, analytical, and functional characteristics of DRL_RI with the reference products (data on file) was done. Following this, a clinical study was conducted in patients with rheumatoid arthritis having inadequate response to methotrexate-based therapy and no prior biologic administration demonstrated a three-way pharmacokinetic (PK) similarity and comparable efficacy, pharmacodynamic (PD), safety, and immunogenicity of DRL_RI with the reference products (Rituxan and MabThera).11,12 Clinical similarity of DRL_RI and reference rituximab was also demonstrated in patients with diffuse large B-cell lymphoma with equivalent PK, and comparable efficacy, PD, safety, and immunogenicity. 11
As part of the continuing clinical development for DRL_RI, this phase III study (RI-01-006; FLINTER) was conducted to demonstrate equivalent efficacy of DRL_RI to EU-approved rituximab (MabThera) in patients with previously untreated Stage II–IV, CD20-positive, LTB-FL. This study also evaluated safety, immunogenicity, rituximab concentrations, and PD of DRL_RI compared with MabThera. This data are reported post study completion (February 27, 2023) when the last randomized patient completed 52 weeks of follow-up.
Methods
We used the CONSORT 2010 reporting parallel group randomized trials. 13
Study design and population
The FLINTER Study was a randomized, double-blind, parallel-group, multicenter, phase III trial (EudraCT number: 2018-004223-36; ClinicalTrials.gov identifier: NCT03976102) in patients with previously untreated, CD20-positive LTB-FL, conducted at 142 centers in 17 countries across the United States, Europe, and Asia (participating countries, sites, and investigators details are provided in Supplemental Material 1). Ethics committee approvals were obtained for each participating center, and all patients provided written informed consent for participation. The study design is presented in Figure 1.

Study design.
Adult male or female patients with Ann Arbor Stage II, III, or IV of histological Grade 1, 2, or 3a previously untreated, CD20-positive, LTB-FL as per Groupe d’Etude des Lymphomes Folliculaires (GELF)-based criteria 14 with at least one measurable lesion were enrolled. Patients had an Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 with a life expectancy >3 months, and a serum lactate dehydrogenase (LDH) level within normal limits at Screening. Prior use of rituximab, any CD20 monoclonal antibodies, or any prior chemotherapy and radiotherapy for FL were excluded. A full list of patient eligibility criteria is provided in Supplemental Material 2. Patients were randomized 1:1 to receive either DRL_RI or MabThera; randomization was stratified by low-, medium-, and high-risk patients using the Follicular Lymphoma International Prognostic Index 2 (FLIPI2), by tumor grade (1–2 vs 3a) and by geographical area. Since the study was conducted during the COVID-19 pandemic, adequate measures recommended by regulatory authorities15,16 were implemented for the safety and well-being of patients; details are included in Supplemental Material 2.
Study treatments
The test product, DRL_RI, manufactured by DRL, and the reference product (MabThera) manufactured by Roche Genentech were supplied as sterile concentrates for solution for infusion either as 100 mg/10 mL or 500 mg/50 mL single-dose vials. Both products were packaged and supplied as blinded investigational products in the external packaging (carton) with a unique container number.
All patients received intravenous infusions of 375 mg/m2 DRL_RI or MabThera weekly for 4 weeks (induction period), followed by maintenance treatment at the same dose every 8 weeks starting at Week 12 until Week 36, and follow-up at Week 52, that is, end of study (EOS). The infusion rate and premedications were in line with MabThera’s prescribing information. 6
Study assessments and end points
Efficacy assessments
The primary efficacy end point was best overall response rate (BORR), defined as proportion of patients in each treatment group that achieved the best overall response of either complete response (CR), unconfirmed complete response (uCR), or partial response (PR) up to Week 28 based on blinded independent central review, in accordance with the response criteria for malignant lymphoma. 17 Disease assessments were scheduled at Screening, Weeks 12, 28, and 52, regardless of treatment delays. All CT or PET-CT imaging data sets were transferred to a blinded independent central imaging vendor for centralized response assessment of primary and other efficacy end points. Secondary efficacy end points included overall response rate (ORR) at Week 12 and Week 28, CR rate at Week 28; duration of response (DOR), progression-free survival (PFS), and overall survival (OS) up to Week 52. The exploratory efficacy end point included ORR based on the Lugano criteria 18 for patients with an available PET Scan.
Safety and immunogenicity assessments
Safety assessments included adverse events (AEs), treatment-emergent adverse events (TEAEs) and serious adverse events (SAEs), infusion-related reactions (IRRs), vital signs and physical examination, clinical safety laboratory tests, 12-lead ECG, and ECOG performance status throughout the study. COVID-19 events were considered Events of Special Interest (EOSIs). AEs were coded using MedDRA Version 25.0, and severity was graded as per the U.S. National Cancer Institute Common Terminology Criteria for Adverse Events version 5.0.
Blood samples for antidrug antibody (ADA) and neutralizing antibody (NAb) were collected prior to study drug infusion on Day 1 and Day 22 (Week 4), and Weeks 8, 28, 44, and 52.
Other exploratory assessments
Blood samples for rituximab concentrations were collected up to Week 12 and for potential differences in PD, including time to depletion and repletion up to Week 52.
Statistical methods
Sample size determination and blinded sample size reestimation
Initially, a sample size of 284 (with 15% dropout) was estimated based on ORR as the primary end point. During the study conduct, in agreement with the U.S. FDA, the primary end point was modified to BORR up to Week 28, with symmetrical equivalence margins of ±0.17. Based on the observed pooled BORR up to Week 28, the revised sample size post blinded sample size reestimation was 312 patients (156 per treatment group). Owing to additional patients already screened, 317 patients were randomized.
Data analysis
The primary analysis was based on the intent-to-treat set (ITT) and included all randomized patients. The per-protocol set (PPS) included all patients who received at least 1 dose of the study drug and had measurable disease at baseline as confirmed by central review, had at least one available valid response evaluation up to Week 28, and had no major protocol deviations impacting the primary end-point.
ITT was used for the primary efficacy end point. Two-sided 90% and 95% confidence intervals (CIs) for the difference in BORR up to Week 28 (DRL_RI—MabThera) were obtained by the unstratified exact score method 19 implemented in SAS Proc FREQ. It was hypothesized that the DRL_RI was equivalent to MabThera applying prespecified symmetrical equivalence margins (±17%). Equivalence was concluded for U.S. FDA and EMA, if the 90% and 95% CIs, respectively, were completely contained within the predefined interval. Multiple preplanned supporting and sensitivity analyses were done using PPS, investigator’s assessments, stratified methods, analysis of estimands for the U.S. FDA and EMA, tipping point analysis, mixed multiple imputation for missing responses, imputation under a non-inferiority assumption, and analysis using study treatment received and stratification data, to further explore the robustness of statistical results; details are included in Supplemental Material 3. ORR, PFS, OS, and DOR by central review were analyzed descriptively and graphically using Kaplan–Meier methods. BORR up to Week 28 from central review based on Lugano criteria 18 was summarized with corresponding 95% CI by treatment group, and concordance between response via Cheson 17 and Lugano criteria 18 was analyzed. Safety was assessed in all patients who received at least 1 dose of the study drug, called the safety analysis set. ADA and NAb results were summarized descriptively, with at least one immunogenicity sample having a valid result. Rituximab concentrations and PD analysis included patients who had at least one available pre-dose sample and one quantifiable study drug concentration. All statistical analyses were conducted using SAS® version 9.4 (SAS Institute, NC, USA).
Clinical trial data sharing
Individual participant data will not be shared.
Results
Patient disposition
Between May 15, 2019 and February 27, 2023, 317 patients were randomized either to DRL_RI (n = 162) or MabThera (n = 155). Of these, 143 (88.3%) patients in DRL_RI group and 129 (83.2%) patients in MabThera group completed the study (Week 52). In all, 19 patients discontinued from DRL_RI group and 26 patients from MabThera group; patient disposition and analysis sets are presented in the CONSORT flowchart (Figure 2).
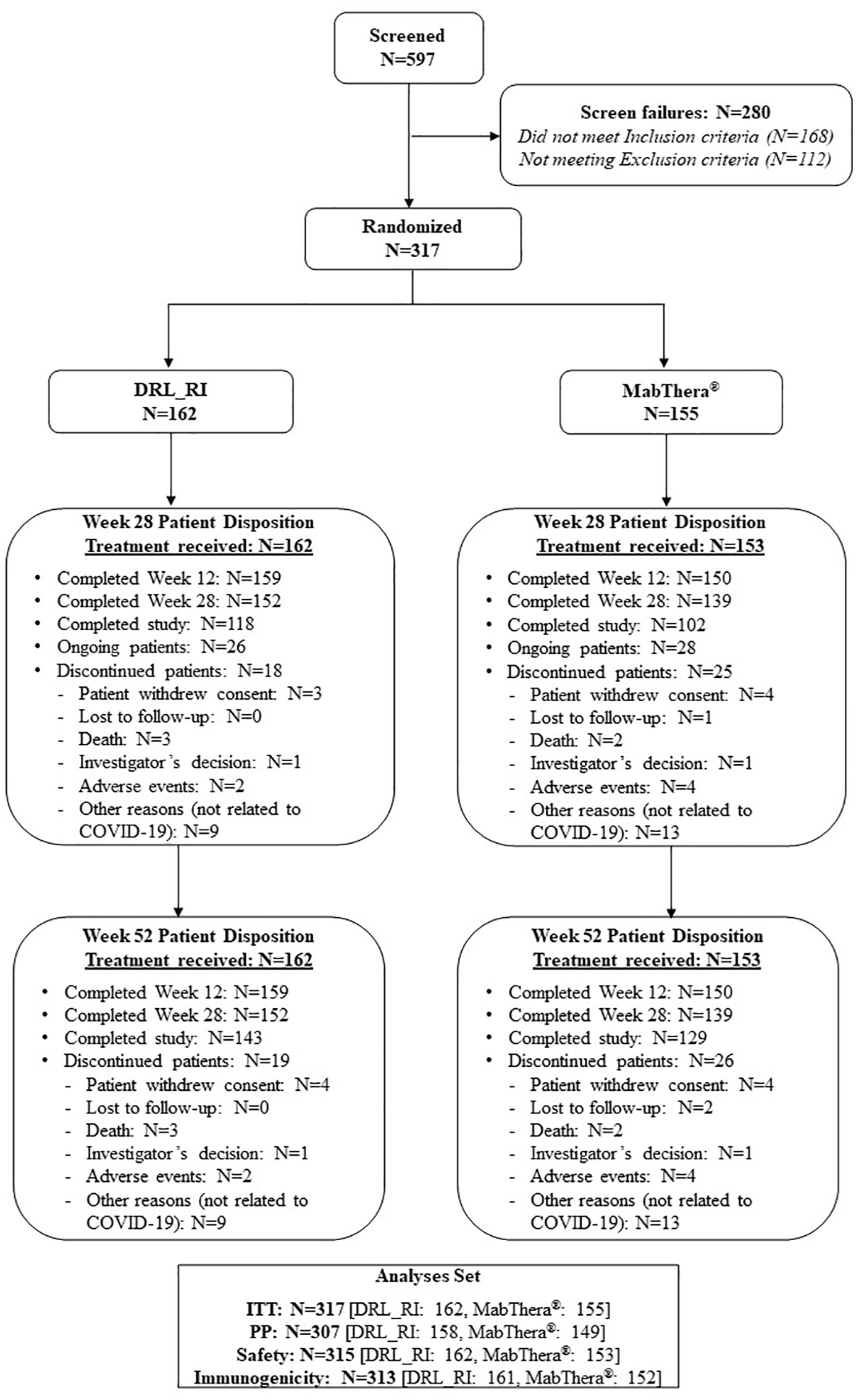
Patient disposition—CONSORT flow chart.
Demographic and baseline characteristics
Demographic and baseline disease characteristics were well balanced between treatment groups (Table 1). The median age for the study group was 58 (22–87) years with a balanced male:female distribution; 66.6% were White. Most patients (78.9%) had an ECOG PS score of “0,” 57.7% had “intermediate risk” as per the overall FLIPI2 risk category; 84.2% were Grade 1–2, and 15.8% were Grade 3a FL. Medical history, concurrent illnesses, prior and concomitant treatments, study treatment compliance, and extent of exposure were comparable between groups. Major protocol deviations due to COVID-19 occurred in 3 (1.9%) patients in DRL_RI group and 6 (3.9%) patients in MabThera group.
Demographic and baseline characteristics (ITT analysis set).

% calculated using the number of patients in the ITT analysis set for each treatment group, or overall study group, as the denominator (n/N * 100).
All BSAs were summarized together regardless of their calculation method.
These two patients in MabThera group were randomized but not dosed, and hence, data were missing.
These two subjects in MabThera arm were randomized but not dosed, and hence, data were missing.
The patients with LDH levels high at Screening were assessed again prior to randomization and assessed by the investigator. For four patients, the LDH levels were above ULN, even at randomization, however, the rise was minimal and clinically not significant as per the investigator’s discretion. Hence, these patients were randomized into the study.
BSA, body surface area; DRL_RI, proposed rituximab biosimilar; ECOG, Eastern Cooperative Oncology Group; eCRF, electronic Case Report Form; EU, European Union; FL, follicular lymphoma; FLIPI2, Follicular Lymphoma International Prognostic Index 2; ITT, intent-to-treat; IWRS, Interactive Web Response System; LDH, lactate dehydrogenase; max, maximum; min, minimum; n, number of patients in specific category; N, number of patients in the ITT analysis set; SD, standard deviation; ULN, upper limit of normal.
Efficacy
Primary efficacy end point
BORR up to Week 28 based on blinded independent central review was 80.25% (95% CI: 73.27–86.08) for DRL_RI and 79.35% (95% CI: 72.12–85.43) for MabThera for the ITT (Table 2), the corresponding difference in BORR was 0.89% (90% CI: −6.67 to 8.48; 95% CI: −8.05 to 9.93). The 90% and 95% CIs for the primary end point were within the prespecified equivalence margin of ±17%. CR as the best response was reported in 34.0% and 35.5%, PR in 39.5% and 32.9%, and uCR in 6.8% and 11.0% of patients in the DRL_RI and MabThera groups, respectively.
Summary of BOR up to week 28 based on central radiology review and primary end point analysis (ITT analysis set).

Percentages were calculated using the number of patients in the ITT analysis set as the denominator (n/N * 100). CIs were calculated using the unstratified exact score method 19 with treatment as an explanatory variable and BOR as a response. Unknown was defined as “not available,” “not evaluable,” or the patient dropped out of the study before a response examination was performed and was considered a nonresponder. The prespecified equivalence margin was ±0.17 (±17%).
The proportion of responders in each treatment group was defined as patients who achieved CR up to Week 28 based on central radiology review in accordance with the response criteria for malignant lymphoma. 17
BORR was defined as the proportion of patients who achieved CR, PR, or complete remission unconfirmed up to Week 28 based on central radiology review in accordance with the response criteria for malignant lymphoma. 17
BOR, best overall response; BORR, best overall response rate; CI, confidence interval; CR, complete response; ITT, intent-to-treat set; n = number of patients in a specific category; N, number of patients in the ITT analysis set; PD, progressive disease; PR, partial response; SD, stable disease; uCR, unconfirmed complete response.
Results of the supporting and sensitivity analyses, including the difference in BORR for the PPS, analyses for the main estimands for U.S. FDA and EMA, and analyses based on the investigator’s assessments were consistent with the primary efficacy analysis (Supplemental Material 4).
Secondary and exploratory efficacy
ORR and CR rates were similar in DRL_RI and MabThera groups (Supplemental Material 4) for the ITT set. ORR (95% CIs) was 75.3% (67.9–81.7) versus 73.5% (65.9–80.3) at Week 28, and 71.6% (64.0–78.4) versus 69.7% (61.8–76.8) at Week 52 for DRL_RI versus MabThera groups. CR rates (95% CIs) were 32.7% (25.6–40.5) versus 34.2% (26.8–42.2) at Week 28, and 43.2% (35.5–51.2) versus 44.5% (36.5–52.7) at Week 52 for DRL_RI versus MabThera groups. Results based on the Investigator’s assessment were similar. The maximum DOR was 49.3 and 43.7 weeks at Week 52 in DRL_RI and MabThera groups, respectively. Estimated 1-year PFS rates (95% CI) of 0.82 (0.75–0.89) versus 0.84 (0.77–0.90), and 1-year OS rates (95% CI) of 0.98 (0.96–1.00) versus 0.99 (0.97–1.00) were comparable between DRL_RI versus MabThera groups. Median values of DOR, PFS, and OS were not evaluable and not interpretable for both groups in the current analysis. A similar proportion of patients with CR, uCR, or PR (5.2% vs 4.7%) died or had disease progression in DRL_RI versus MabThera groups.
At Week 28, 158 (49.8%) patients in the ITT set were evaluable for assessment by Lugano 18 criteria (Table 4). BORR based on Lugano criteria 18 was 75.7% (95% CI: 64.3–84.9) for DRL_RI versus 73.8% (95% CI: 63.1–82.8) for MabThera group (Table 3). Overall, 14.9% of patients in DRL_RI group and 15.5% of patients in MabThera group were classified as “responders” by Cheson 17 but as “nonresponders” by Lugano 18 criteria. Additionally, 6.8% patients in DRL_RI and 4.8% of patients in MabThera groups were classified as “nonresponders” by Cheson 17 but as “responders” by Lugano 18 criteria. In all, 79.1% of patients had concordance for BORR at Week 28 between the Lugano 18 and Cheson 17 criteria (primary end point) (Supplemental Material 4).
Summary of BORR by Lugano criteria (Lugano 2014) at week 28 based on central radiology review (ITT).

Percentages were calculated using the number of patients in the ITT analysis set with available PET as the denominator (n/N * 100). The 95% CI was calculated using the exact binomial method with treatment as an explanatory variable and BORR as a response.
The proportion of responders in each treatment group is defined as patients who achieved CR or PR. The BORR was determined by using tumor response based on Lugano criteria 18 only for those patients with available PET data.
BORR, best overall response rate; CI, confidence interval; CR, complete response; ITT, intent to treat set; n, number of patients in a specific category; N, number of patients in the ITT analysis set with available PET; PD, progressive disease; PET, positron emission tomography; PR, partial response; SD, stable disease.
Overview of adverse events (safety analysis set).

Percentages were calculated using the number of patients in the safety analysis set as the denominator (n/N * 100).
Three deaths were Grade 5 severity and two deaths were not graded as Grade 5, however, the outcome of the TEAEs reported was death: One patient in the DRL_RI group died due to a Grade 4 AE of COVID-19 and another patient in the MabThera group died due to a Grade 3 AE of pneumonia.
AE, adverse event; CTCAE, Common Terminology Criteria for Adverse Events; DRL_RI, rituximab biosimilar; E, number of events; n, number of patients in specific category; N, number of patients in the safety analysis set; TEAE, treatment-emergent adverse event.
Subject may be counted in more than one category if AEs in more than one category were reported by the subject.
Safety
The incidence of TEAEs was comparable between groups: 69.8% in DRL_RI and 67.3% in MabThera (Table 5). Most TEAEs were Grade 1 (50.5%) and Grade 2 (34.9%). The most frequently reported TEAEs (Table 5) were COVID-19 (13.6% in DRL_RI vs 13.1% in MabThera groups) followed by pruritus (6.8% DRL_RI vs 6.5% in MabThera groups). About 30% of TEAEs in both groups were drug related; pruritus was the most common drug-related TEAE (5.6% in DRL_RI vs 4.6% in MabThera groups). Grade 4 drug-related TEAEs were neutropenia (one patient in DRL_RI and two patients in MabThera groups), neutrophil count decreased (two patients in MabThera group), and muscular weakness (one patient in MabThera group).
Summary of TEAEs (occurring in ⩾5% patients) in the safety analysis set.
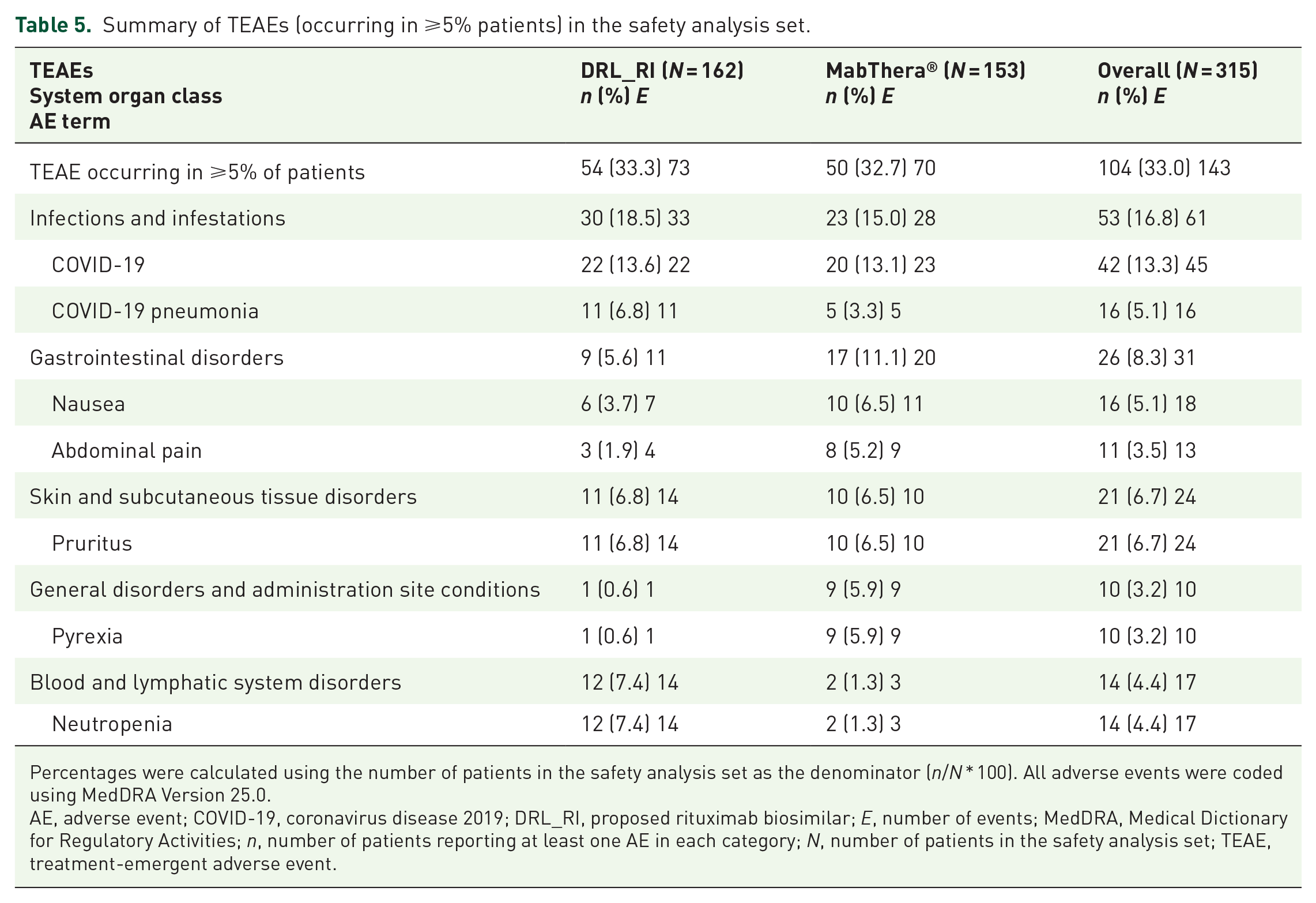
Percentages were calculated using the number of patients in the safety analysis set as the denominator (n/N * 100). All adverse events were coded using MedDRA Version 25.0.
AE, adverse event; COVID-19, coronavirus disease 2019; DRL_RI, proposed rituximab biosimilar; E, number of events; MedDRA, Medical Dictionary for Regulatory Activities; n, number of patients reporting at least one AE in each category; N, number of patients in the safety analysis set; TEAE, treatment-emergent adverse event.
Five deaths (1.6%) were reported; none were drug related. Three (1.9%) deaths in DRL_RI group were due to COVID-19 pneumonia (1), COVID-19 (2); while two (1.3%) deaths in MabThera group were due to COVID-19 pneumonia, and community-acquired left-sided lower lobe pneumonitis. Serious TEAEs occurred in 13.6% and 13.7% of patients in DRL_RI versus 13.7% MabThera groups; COVID-19 pneumonia was the most common SAE (6.2% in DRL_RI and 3.3% in MabThera). All drug-related SAEs (6 (1.9%)) were Grade 3/4 and included two (1.2%) events (IRRs and neutropenia) in DRL_RI group and four (2.6%) events in MabThera group (anal abscess, COVID-19 pneumonia, muscular weakness, and psychomotor hyperactivity).
IRRs occurred in 28 (17.3%) patients in DRL_RI group versus 32 (20.9%) patients in MabThera group, and mostly included skin and subcutaneous tissue disorders (8.6% vs 8.5%) and respiratory, thoracic, and mediastinal disorders (5.6% vs 4.6%). Pruritus (4.9% vs 4.6%) and throat irritation (4.3% vs 2.0%) were the most common IRRs. The most frequent treatment-emergent EOSI was COVID-19 (13.6% in DRL_RI vs 13.1% in MabThera group).
Two patients (1.2%) were discontinued from DRL_RI due to COVID-19 pneumonia and angina unstable; not drug related. Five patients (3.3%) were discontinued from MabThera, three due to drug-related TEAEs (COVID-19 pneumonia, psychomotor hyperactivity, pneumonia), and two due to TEAEs which were not drug related (thyroid cancer and squamous cell carcinoma). Details of SAEs, drug-related TEAEs, TEAEs leading to discontinuation, and EOSIs are provided in Supplemental Material 5.
Immunogenicity
Eleven (3.5%) patients—eight (5.0%) in DRL_RI group and three (2.0%) in MabThera group—had at least one positive ADA result posttreatment till Week 52. Three of the eight patients in DRL_RI group continued to test positive at EOS of which, two patients tested positive for NAb, one at EOS, and the other at Weeks 8, 28, 44, 52 (EOS). Two of the three ADA-positive patients in MabThera group continued to test positive at Week 52; none tested positive for NAb. A full detail of NAb and ADA assay methodology is provided in Supplemental Material 2.
Trough concentrations of rituximab till week 12
Rituximab concentrations were evaluable in 89 patients (DRL_RI: 48, MabThera: 41). Mean pre-dose concentrations were 66.270 and 68.219 µg/mL in DRL_RI and MabThera groups, respectively, which increased to 288.636 and 298.595 µg/mL, respectively, prior to the end of infusion at Week 2. Mean concentrations continued to increase till prior to the end of infusion at Week 4—415.436 and 417.463 µg/mL in DRL_RI and MabThera groups, respectively, and declined thereafter. At Week 12, the mean concentrations were 248.233 and 286.518 µg/mL in DRL_RI and MabThera groups. The details of PK assay methodology are provided in Supplemental Material 2.
Pharmacodynamics
PD results were reported for 85 patients (DRL_RI: 43, MabThera: 41). Peripheral B-cell counts declined below quantifiable limits following completion of the first dose for most patients in both groups and were also nonquantifiable prior to initiation of Week 2 infusion. Prior to Week 52 infusion, 25 patients in DRL_RI group and 28 patients in MabThera group reported nonquantifiable B-cell counts. The median time to B-cell depletion was 6.9 and 7.0 days for DRL_RI and MabThera groups, respectively. The details of PD assay methodology are provided in Supplemental Material 2.
Discussion
Efficacy equivalence, as assessed by BORR, was demonstrated for DRL_RI and MabThera administered as a monotherapy in LTB-FL. The 90% and 95% CIs for BORR difference up to 28 weeks between treatments were well within the prespecified equivalence margin of ±17%. All secondary and sensitivity analyses supported the primary analysis as per the U.S. FDA (based on 90% CI) and EMA (based on 95% CI) criteria. DRL_RI was well tolerated with an overall safety and immunogenicity profile comparable to MabThera. 6
A similar phase III study 20 had confirmed therapeutic equivalence of a rituximab biosimilar with 90% CIs within the prespecified margin of ±17%. The margin used and the results obtained in our study are aligned with similar randomized studies that tested biosimilars’ equivalence with rituximab.20–22 Comparing the PK, efficacy, and safety including immunogenicity of a proposed biosimilar to a reference product is an essential component of a clinical development program providing evidence for biosimilarity. Previous study RI-01-003 concluded three-way PK equivalence of DRL_RI with both reference products (MabThera and Rituxan) with 91% CI for the test-to-reference ratios of all primary and secondary PK end points within the prespecified margins of 80.00% to 125.00%. 12 The current data, together with other studies11,12 of DRL_RI and rituximab reference products, establish the similarity of DRL_RI with reference (Rituxan, MabThera) for efficacy, safety, PK, PD, and immunogenicity end points.
In this study, efficacy equivalence was demonstrated for DRL_RI and MabThera in patients with CD20 positive, LTB-FL as a first-line treatment. LTB-FL is a slow-growing malignancy, and rituximab monotherapy is an acceptable option according to treatment guidelines7,8 based on studies of rituximab monotherapy associated with a high response rate and low toxicity.23–25 Immediate treatment with rituximab monotherapy is shown to significantly delay disease progression and the time to chemotherapy/radiotherapy over a watch-and-wait approach in asymptomatic LTB-FL, suggesting rituximab monotherapy as a standard approach for managing these patients. 24 Previously untreated, CD20-positive LTB-FL patients represent the most suitable patient pool as they present with more uniform disease characteristics, providing a more sensitive model to assess efficacy equivalence without any confounding factors, against the heterogeneous NHL subtypes that need rituximab to be administered in combination with chemotherapy.20,21,26 BORR as an end point also maximized sensitivity to differences in a reasonable time in this population, where the time to tumor growth was prolonged. Low tumor burden was assessed using GELF-based criteria,14,26 and normal serum LDH was a prerequisite for enrollment, as LDH is an important prognostic factor in FL. A dose of 375 mg/m2 was maintained to allow a comparative evaluation of DRL_RI’s safety and efficacy in line with MabThera’s prescribing information, 6 and the most common treatment schedule of 4 doses weekly and 4 doses every 2 months was chosen. All these measures imply the robustness of this study’s design.
The BORR difference up to Week 28 of 0.89% (90% CI: −6.67 to 8.48; 95% CI: −8.05 to 9.93) was entirely within the prespecified margin (±17%), and 79.1% patients had a concordance between Cheson 17 and Lugano 18 criteria for BORR. CR rates as a best response were 34.0% in DRL_RI versus 35.5% in MabThera groups. While median DOR, PFS, and OS were not interpretable, which might be attributed to the slowly progressing nature of the disease, overall secondary end points were comparable between groups. Overall, the results from this study are comparable to studies of other rituximab biosimilars.20–22 Ogura et al. 20 reported an ORR of 83% with CT-P10 and 81% with rituximab-EU, with a 1.8% difference by month 7. The JASMINE study 22 that compared biosimilar ABP 798 with rituximab in FL reported a BORR of 78.0% in ABP 798 and 70.2% in rituximab groups, while another study 21 of rituximab biosimilar PF-05280586 and rituximab-EU as induction therapy reported an ORR of 75.5% versus 70.7% at Week 26 with a difference of 4.66%. Further, a recent meta-analysis 24 of four randomized studies (ClinicalTrials.gov identifier: NCT02809053)20–22 of rituximab biosimilars and MabThera reported no significant difference in the overall ORR between the biosimilars and rituximab (relative risk = 1.00, 95% CI: 0.93–1.08, p = 0.92) for the treatment of LTB-FL. The dose regimen and treatment schedule in our study are the same as the study by Ardeshna et al. 24 that evaluated the use of single-agent rituximab in patients with LTB-FL across three arms (rituximab induction, wait-and-watch, rituximab maintenance). Significantly more overall responses were reported in the maintenance group (91% and 84%) as compared to the induction group (77% and 57%) at month 7 and month 25. In the study by Ardeshana, 56% patients in the watchful waiting group versus 17% in the maintenance group required a new treatment owing to disease progression. 24 The induction regimen in our study is also similar to that of RESORT trial, wherein 70.8% (95% CI: 67%–76%) patients with LTB-FL responded to the single-agent rituximab induction therapy of 4 weeks. 25
Rituximab concentrations at different time points and PD parameters were comparable between DRL_RI and MabThera. The rapid peripheral B-cell depletion following completion of the first dose observed in both groups is similar to the earlier study of DRL_RI in rheumatoid arthritis. 12 These findings are also aligned with the phase III study of biosimilar CT-P10 versus reference rituximab, indicating similarity in the extent of B-cell depletion. 20 In light of this literature, the efficacy equivalence demonstrated by DRL_RI in this study supports its biosimilarity.
The overall AE profile of DRL_RI was comparable to rituximab, 6 with no new, unexpected safety findings. About 30% of TEAEs were considered drug related, of which, IRRs were 17.3% in DRL_RI and 20.9% in MabThera; pruritus and throat irritation being the most common reactions. Overall, AEs related to infections and IRRs were less common in this study than in other innovator rituximab registration studies that report ⩾30% to 55% infections in patients with NHL and >50% IRRs in patients with NHL or CLL.5,6,20,26 The drug-related SAEs of IRR and neutropenia with DRL_RI are similar to the serious TEAEs experienced with rituximab during the monotherapy study. 24 There were no significant differences in safety between biosimilars and rituximab from the meta-analysis; the relative risk for SAEs was 1.15 (95% CI: 0.69–1.89, p = 0.59) and for IRRs was 0.91 (95% CI: 0.77–1.09, p = 0.32) from this pooled data, 27 demonstrating the comparable safety of rituximab biosimilars in treatment of LTB-FL. The EOSIs, COVID-19 occurrence was 19.1% versus 15.0% in DRL_RI versus MabThera. COVID-19 was the most frequently reported TEAEs apart from IRR, and also the underlying cause of four out of five deaths in this study. Except for one EOSI of COVID-19 pneumonia related to MabThera, none were considered drug related, overall negating the impact of COVID-19 pandemic on these results.
A minority of patients (eight in DRL_RI group and three in MabThera group) were ADA positive; of which two patients in DRL_RI group were NAb positive versus none on MabThera. Considering the low incidence of ADA and NAb positivity and its limited influence on efficacy and safety parameters, these findings did not indicate relevant clinical differences between DRL_RI and MabThera groups. The findings concur with a similar phase III trial that reported an ADA incidence of 1%–2% for the biosimilar CT-P10 and rituximab in similar patients. 20 The low ADA incidence is also comparable to the reported incidence of 1%–56% across indications in other rituximab registration studies.5,6
Rituximab biosimilars demonstrating comparable efficacy and safety represent a potential therapeutic alternative to rituximab therapies for multiple indications, which otherwise pose access and affordability concerns to patients across geographies. Systematic evaluations and approvals of biosimilars such as DRL_RI may reduce the economic burden and offer increased affordability and accessibility to improve patient treatment and outcomes with an equivalent efficacy and safety to the reference products.
Conclusion
This study demonstrated efficacy equivalence of DRL_RI with MabThera in previously untreated CD20-positive LTB-FL patients. DRL_RI was well tolerated with safety, immunogenicity, serum concentrations, and PD profiles comparable to MabThera.
Supplemental Material
sj-pdf-1-tam-10.1177_17588359251339925 – Supplemental material for Efficacy and safety of rituximab biosimilar (DRL_RI) versus MabThera® in low-tumor-burden follicular lymphoma: the FLINTER study
Supplemental material, sj-pdf-1-tam-10.1177_17588359251339925 for Efficacy and safety of rituximab biosimilar (DRL_RI) versus MabThera® in low-tumor-burden follicular lymphoma: the FLINTER study by Narendra Maharaj, Dharma Rao Uppada, Anand Eswaraiah, Ranjith Kakkattu, Pramod Reddy, Volha A. Kalenik, David Belada, Ana Oliveira Ramos, Jin Seok Kim and Yauheni V. Baranau in Therapeutic Advances in Medical Oncology
Footnotes
Acknowledgements
The authors acknowledge the contributions from Dr. Luis Lopez Lazaro, Dr. Suresh Kankanwadi (former DRL employees) for the study conception, Dr. Ankit Ranpura (former DRL employee), Dr. Vikas Kumar, Dr. Naveen Reddy, Dr. Kamala Bhavaraju, Debashish Dutta (DRL employees) for their contributions to the study conduct and enthusiastic collaboration of all the participating investigators at the study centers. The authors also thank Dr. Auro Viswabandya, who acted as Data Monitoring Committee Chair, along with members Dr. Reena Nair and Ms Jaishree Ganjiwale. The authors thank clinical science team Dr. Tazeen Aamena Idris, Dr. Routhu Kasi Viswanath (from Dr. Reddy’s Laboratories Ltd) and Dr. Hetal Shah from MeWriT Healthcare Consulting, Ahmedabad for providing writing assistance.
Author’s note
A complete list of the participating centers and investigators appears as a data supplement to the online version of this article.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.