
Abstract
Immune checkpoint blockade has modified the treatment landscape for many types of tumors, including lung cancer. Still our knowledge on the biology of the interaction between tumor cells and the microenvironment is limited, preventing the optimal use of these new compounds and the maximum benefit that the patients can derive from them. We have actively worked on the role of STAT3, a transcriptional factor that causes innate resistance to targeted therapies in oncogene-addicted tumors. In this short review we take the opportunity to express our opinion and review existing knowledge on the immune role of STAT3 and the possible implications that this may have for the discovery of new biomarkers to predict response to immunotherapy, as well as new partners to combine with and increase the efficacy of immune checkpoint inhibitors.
Introduction
Immune checkpoint inhibitors, like anti-programmed cell death-1 (PD-1) or anti-programmed death-ligand 1 (PD-L1) antibodies, have demonstrated positive results in non-small cell lung cancer (NSCLC) patients, with prolonged overall survival and durable responses. 1 Nevertheless, the response rate to immunotherapy does not exceed 30% in the second-line setting,2–4 or 45% in the first-line setting. 5 Tumor recurrences are common clinical findings, and disease progression can occur even after a durable response, but there is little knowledge about the complex mechanisms of resistance to immune checkpoint inhibitors. 6 Surprisingly, the T-cell activation through immune checkpoint blockade may trigger the proliferation of cancer cells in certain tumor types that are dependent on T cells.7,8 Therefore, greater effort should be made in order to raise the bar of efficacy and identify patients who can substantially benefit from therapies targeting the immune system. Combination strategies appear to be an interesting approach to optimize immunotherapy treatment outcome in NSCLC patients.9,10 An example is the combination of chemotherapy with the anti-PD-1 antibody, pembrolizumab, that has been approved as first-line treatment in patients with lung adenocarcinoma. 11 Many clinical trials combining anti-PD-1/PD-L1 antibodies with other immune checkpoint inhibitors are also ongoing. 12
The discovery of predictive biomarkers is necessary for improving the efficacy of immunotherapy. Until now, great attention has been paid to the expression of PD-L1 as a predictive marker of response to immune checkpoint blockade.2,3,5,13 Immunohistochemistry evaluation of PD-L1 expression is the only available biomarker in the clinical setting, but its role is still controversial. High tumor mutational burden is also related to the outcome of immune checkpoint blockade.14,15 With the exception of KRAS and BRAF-mutant NSCLC, other lung adenocarcinomas driven by oncogenic alterations, like epidermal growth factor receptor (EGFR) mutations or echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase (EML4-ALK) fusions, have low mutational burden 16 and seem to derive minimal benefit from immunotherapy. 17
We and others have highlighted the role of signal transducer and activator of transcription 3 (STAT3) and yes-associated protein 1 (YAP1)-Src in innate resistance to EGFR tyrosine kinase inhibitors (TKIs) in EGFR-mutant NSCLC.18–22 We are currently defining the regulatory role of STAT3 and YAP1-Src on the expression and activation of receptor tyrosine kinases (RTKs) in EGFR mutants, 23 as well as other oncogene-addicted tumors. 24 In this short review we will summarize the available data on STAT3 and PD-L1 and provide a rationale for STAT3 as a potential biomarker as well as a target for optimizing treatment with immune checkpoint inhibitors.
STAT3 signaling and antitumor immunity
It is well recognized that STAT3 plays a critical role in cancer.25–27 It also affects many aspects of the immune system. 28 STAT3 induces PD-L1 up-regulation in many tumors and therefore assists cancer cells to escape immune surveillance.29,30 It binds directly to the promoter of PD-L1, both in antigen-presenting cells 31 and in tumor cells. 32 It also activates DNA methyltransferase 1 (DNMT1), which methylates the promoter region, and subsequently suppresses the expression of immunoproteasome (PSM) subunits and major histocompatibility complex (MHC) molecules. In contrast, STAT1, which is negatively regulated by STAT3, induces the expression of PSM subunits and MHC molecules 33 (Figure 1). PSMs generate peptides on the cell surface that fit in the groove of MHC molecules and allow surveillance by CD8 T cells. 34 PSMB8 and PSMB9 expression is higher in EGFR-mutant than in EGFR-wildtype NSCLC cell lines, suggesting a stronger role of STAT3 in immune suppression in EGFR-mutant lung cancer. 33
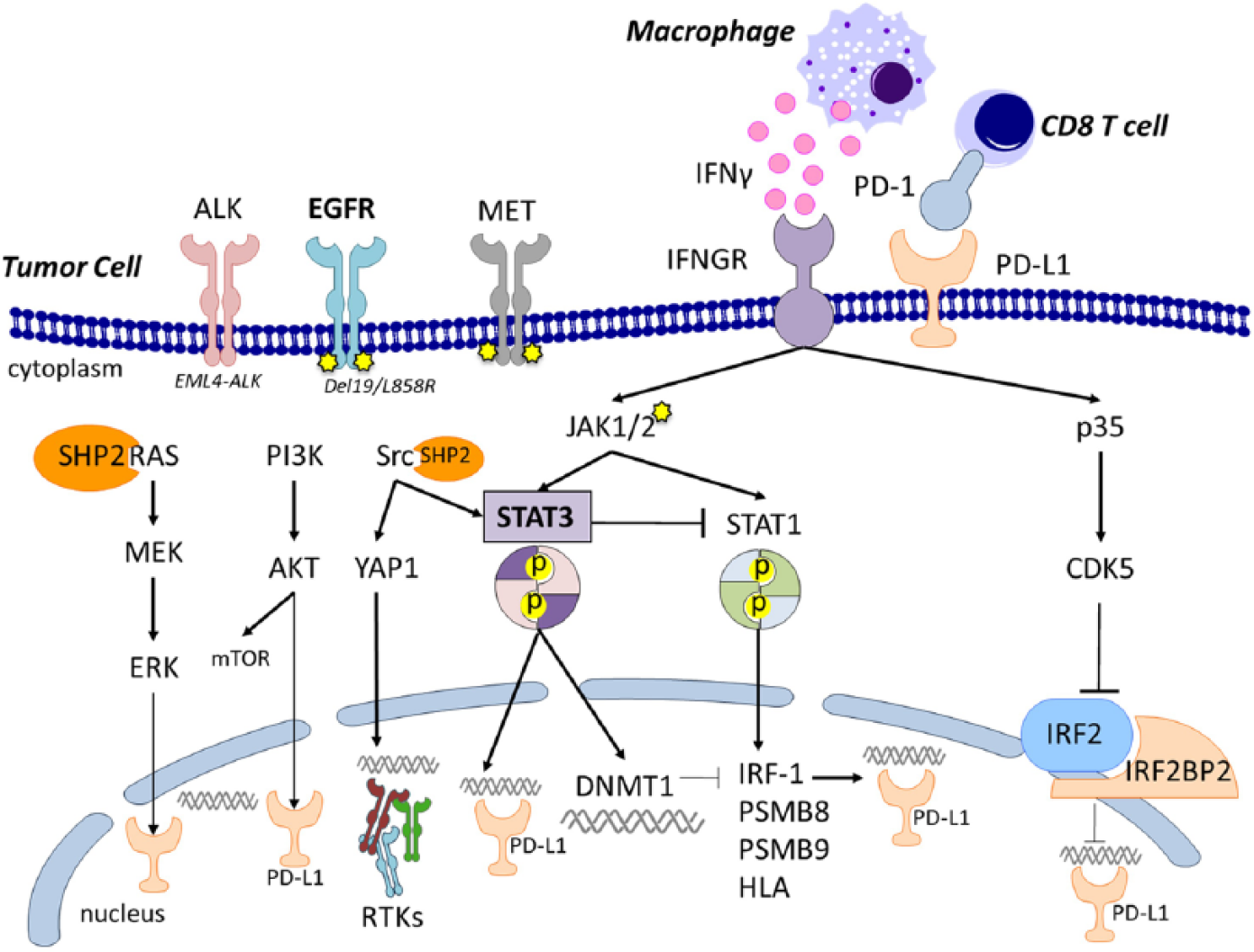
Programmed death-ligand 1 (PD-L1) is the ligand of programmed death 1 (PD-1) receptor. PD-1 is expressed on the T-cell surface (mainly CD8+ T-lymphocytes), while PD-L1 is presented on the cell surface by antigen-presenting cells (APCs; as macrophages) and tumor cells. Epidermal growth factor receptor (EGFR)-activating mutations are located in the tyrosine kinase domains and mainly in the form of a base-pair deletion at exon 19 (ΔE746_A750) or a point mutation at exon 21 (L858R). Anaplastic lymphoma kinase (ALK) rearrangements involve gene fusion partners, leading to constitutive protein activation. In NSCLC the echinoderm microtubule-associated protein-like 4 (EML4)-ALK variant is the most frequently reported fusion gene. The MET receptor can be hyperactivated through gene amplification (copy number gain), or gene splicing variants. EGFR, ALK and MET signal via tyrosine phosphorylation lead to the activation of mitogen-activated protein kinase (MAPK), signal transducer and activator of transcription 3 (STAT3) and AKT pathways. MAPK pathway is mainly dependent on Src homology 2 domain-containing phosphotyrosine phosphatase 2 (SHP2). SHP2 interferes with the process of Ras inactivation catalyzed by Ras GTPase-activating protein (RasGAP), hence increasing the half-life of activated Ras (GTP-Ras). MAPK/ERK Kinase 1 (MEK1) and MEK2 are activated upon phosphorylation by Ras cascade. MEK1/2 is in turn able to phosphorylate and activate extracellular signal-regulated kinase (ERK1 and ERK2). AKT is the major downstream target of phosphatidylinositol 3-kinase (PI3K), which is activated by the receptor tyrosine kinases (RTKs). Activated PI3K induces the recruitment of AKT to the cell membrane, driving a conformational change in the protein. This enables full activation of AKT upon phosphorylation. Following activation, AKT translocates to the cytoplasm and nucleus, and phosphorylates various downstream substrates including mammalian target of rapamycin (mTOR). Both ERK and AKT can induce up-regulation of PD-L1. The specific mechanisms of this activation pathway are still not clearly defined. SHP2 activates several Src family kinase (SFKs), including Src itself. Upon Src activation, several downstream Src binding partners are targeted for phosphorylation, including yes-associated protein 1 (YAP1). YAP1 is a transcriptional coactivator, and has been reported to bind several DNA-binding transcription factors, thus mediating up-regulation of several RTKs. Signaling transducer and activator of transcription 3 (STAT3) can be activated through Janus-like kinase (JAK) only in the presence of Src kinase activity, in low STAT1 conditions. When activated, STAT3 undergoes phosphorylation-induced homodimerization. The homodimer translocates to the nucleus and binds to DNA. Through the activation of DNA methyltransferase 1 (DNMT1), STAT3 downregulates the transcription of several genes involved in immune surveillance: interferon regulatory factor 1 (IRF-1), immunoproteasome subunits (PSM) B8 and B9 and the human leukocyte antigens (HLA). IRF-1 is a PD-L1 inducer; PSMB8-9 and HLA are mediators of effector immune cells activation. STAT1, another member of the STAT family, is activated by JAK upon phosphorylation into dimer conformation and translocates to the nucleus as well. STAT1 has the opposite function of inducing the transcription of IRF-1, PSMB8-9 and HLA. In turn STAT3 can inhibit STAT1 activity. Interferon gamma (IFNγ) is a cytokine released in the tumor microenvironment by the cells of the immune infiltrate. Once binding to its receptor (IFNGR), it triggers signaling cascades that are able to modulate PD-L1 expression. IFNGR can activate both STAT1 and STAT3 through JAK1/2. IFNγ-mediated STAT3 activation also requires SFKs. The alternative activation of either STAT1 or STAT3 is competitive and related to their relative abundance in the tumor cell. IFNGR can also activate cyclin-dependent kinase 5 activator 1 (CDK5R1 or p35), the main CDK5 activator. CDK5 inhibits interferon regulator factor 2 (IRF-2) and IRF2BP2 that are PD-L1 co-repressors.
STAT3 is phosphorylated at tyrosine 705 position through Janus kinase 1/2 (JAK1/2). This phosphorylation mediates the formation of STAT3-STAT3 homodimers, which translocate to the nucleus and bind to DNA promoting oncogenic functions in tumor cells.35,36 In the nucleus, the phosphorylation at serine 727 is mediated by TC45 (protein tyrosine phosphatase nonreceptor type 2, PTPN2) and inactivates pY705. 37 Conversely, the activity of STAT3 in the immune cells is also regulated by dual specificity phosphatase 2 (DUSP2). 38 It seems that naïve CD4-positive T cells undergo T-helper 17 (Th17) differentiation independently of JAK activity. 39 Moreover, Th17 are negatively regulated by DUSP2 activation and subsequent STAT3 dephosphorylation, 38 suggesting a role of STAT3 activation in promoting Th17 differentiation which is dependent on DUSP2 rather than on JAK. Surprisingly, but in line with the concept of alternative mechanisms of action in immune or nonimmune tumor cells, the proliferation signal is driven by p-S727, and not p-Y705, in chronic lymphocytic leukemia cells. 40
Thus, STAT3 has been implicated in regulating the tumor microenvironment through several mechanisms, including the recruitment of myeloid-derived suppressor cells (MDSCs) or the decrease of immune cell infiltration, in different types of tumors.41–46 Tumor-derived factors can activate JAK/STAT3 signaling in hematopoietic cells, leading to abnormal differentiation of dendritic cells, which are thus unable to activate CD8 T cells. On the other hand, abnormal dendritic cells can promote T-cell tolerance and immunosuppressive immature myeloid cells.47–49 Of note, they show increased PD-L1 expression. 49 This mechanism of STAT3 mediated dysfunction of dendritic cells was also found in a NSCLC model. 50
The role of interferon γ
Interferon γ (IFNγ), encoded by the IFNG gene, is secreted by macrophages and other immune infiltrating cells 51 and promotes PD-L1 expression. 10 We have reported that high baseline IFNG mRNA levels can be related to better outcome of NSCLC and melanoma patients treated with the anti-PD-1 antibodies, nivolumab or pembrolizumab, respectively. 52 IFNγ binds to its receptor and activates STAT1 through JAK1/2, but it can also activate STAT3 with opposite biological effect.51,53 IFNγ−mediated STAT3 activation requires Src activity and is weaker in wildtype STAT1 expressing cells than in STAT1-null. 53 The relative abundance of these two members of STAT family in tumor cells can justify the different IFNγ-mediated activation patterns. IFNγ requires cyclin-dependent kinase 5 (CDK5) and its activator, p35, to induce the expression of PD-L1.54,55 When CDK5 is lost, the IFN regulatory factors IRF2 and IRF2 binding protein 2 (IRF2BP2) persist and negatively regulate PD-L1 expression54,55 (Figure 1). Activated STAT1 induces the expression of IRF1, which is a PD-L1 positive regulator. 56 JAK1/2 mutations have been described as a negative predictive biomarker of response to immune checkpoint blockade, since they prevent the IFNγ-mediated up-regulation of PD-L1. 57 In another study, JAK1/2 mutations were not related to nivolumab resistance, 28 which increases the complexity of cancer immune escape. This could be explained by the fact that, in the absence of IFNγ, CKLF-like MARVEL transmembrane domain-containing protein 6 (CMTM6) is a regulator of PD-L1 expression. 58 CMTM6 can be overexpressed in several types of tumors, including lung cancer. 59
STAT3 as an immune modulator in oncogene-addicted tumors
We have shown that early STAT3 activation occurs in EGFR-mutant lung cancer cell lines upon treatment with gefitinib, afatinib or osimertinib, when the drugs are used at 50% of their inhibitory concentration (IC50).18,22 We have also found that combined EGFR and STAT3 inhibition can be more effective than EGFR inhibition alone in treatment-naïve and resistant EGFR-mutant NSCLC cell models, in vitro and in vivo. Moreover, we defined baseline STAT3 mRNA expression as a predictor of outcome to EGFR TKI treatment in EGFR-mutant NSCLC patients. 18 JAK/STAT3 signaling is a common downstream pathway for many RTKs. In addition, c-Src has an important activity in regulating STAT3 function. 60 The increased STAT3 activity in EGFR TKI-resistant cells could be mediated by the co-expression of RTKs, other than EGFR. 61 In castration-resistant prostate cancer, the activation of RTKs, such as, EGFR, AXL (AXL receptor tyrosine kinase) or platelet-derived growth factor receptor a (PDGFRa), could only be abrogated whereas interleukin 1 receptor antagonist (IL-1ra) could only be induced when immune checkpoint blockade was combined with the multi-kinase inhibitor cabozantinib. 62 IL-1ra reduces MDSCs and increases macrophage polarization. 62 This synergistic effect can be explained by the multi-kinase inhibitor-mediated suppression of STAT3, facilitating the activity of the immunotherapy. Consistently, there is evidence that STAT3 inhibition is able to reduce MDSC activity in different cancer types.63,64 So far there is little evidence, however, on the correlation of STAT3 and immune escape in oncogene-driven tumors. In a melanoma model, STAT3 was found to be mediating the up-regulation of PD-L1 occurring in BRAF inhibitor-resistant cells. 65
The expression of PD-L1 in EGFR-mutant or ALK-translocated NSCLC has been widely explored at the preclinical level, as well as in clinical trials. Activated EGFR and ALK induce the expression of PD-L1 in cell line models and cause apoptosis of T cells.66,67 Jiang and colleagues explored the correlation between PD-L1 expression and oncogenic alterations such as KRAS, EGFR, MET, ROS1 or ALK, in NSCLC. 68 PD-L1 protein expression was assessed with the rabbit monoclonal anti-human PD-L1 antibody, clone E1L3N (Cell Signaling Technology, Inc., Denver, MA, USA).68,69 They reported that, in lung adenocarcinoma, when using a cutoff of ⩾5% for PD-L1 positivity, there was a 76% overlap between PD-L1 positivity and the presence of oncogenic alterations. The overlap was 67% when the PD-L1 cutoff was ⩾50%. 68 Overall, they found that PD-L1 was expressed only in 36.5% and 12.8% of total lung adenocarcinoma samples, when using a cutoff of ⩾5% and ⩾50%, respectively, 68 consistent with the findings of the main clinical trials.5,11,70 In the first cohort of the ATLANTIC phase II clinical trial, 72.5% of EGFR-mutant or ALK-translocated patients had more than 25% PD-L1 expression. 71 These data indicate that oncogene-addicted NSCLCs have higher PD-L1 expression than those not driven by an oncogenic alteration.
We recently demonstrated that high PD-L1 mRNA expression is significantly related to better progression-free survival in EGFR-mutant NSCLC treated mainly with gefitinib, 72 D’Incecco and colleagues previously reported similar data, with PD-L1-positive EGFR-mutant NSCLC patients experiencing better responses to gefitinib or erlotinib in comparison with PD-L1-negative patients.73,74 In contrast with EGFR-mutant NSCLC, the predictive role of PD-L1 expression in ALK-translocated tumors is not clear. 75 Either EGFR or ALK inhibitors down-regulate PD-L1 expression in EGFR-mutant or ALK-translocated human lung cancer cell lines.66,67 No synergistic effect was found when EGFR or ALK inhibitors were combined with anti-PD-1/PD-L1 antibodies in co-culture models. Since STAT3 levels can be regulated by RTKs other than EGFR or ALK, we can speculate that the absence of synergism in those models is dependent on STAT3 activation. At the time of resistance to EGFR or ALK inhibitors, the PD-L1 expression is raised again.66,67 EGFR-mutant NSCLC cell lines with MET-mediated acquired resistance to EGFR TKIs, had concomitant up-regulation of PD-L1, which was decreased after treatment with a MET inhibitor. 76 Considering that the signaling pathways downstream of RTKs are common, we speculate that there is a similar immune modulation pattern in these two subgroups of NSCLC (Figure 1).
In EGFR-mutant NSCLC, STAT3 is directly related to the expression of PD-L1. Specifically, inhibition or silencing of STAT3, or inhibition or knockdown of the driver mutation is able to down-regulate PD-L1 expression in EGFR-mutant or ALK-translocated lung cancer cell lines.75,77 However, all three main oncogenic signaling pathways, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)/AKT and STAT3 can be involved in PD-L1 expression. Chen and colleagues have shown that activated EGFR-induced PD-L1 expression was abrogated with a MAPK inhibitor, but not with an AKT inhibitor. 66 In an EGFR-mutant NSCLC cell line with acquired resistance to gefitinib, PD-L1 expression was down-regulated after STAT3 or AKT, but not after MAPK inhibition. 78 An increase in STAT3 and AKT phosphorylation, as well as PD-L1 expression, was observed in KRAS-mutant lung cancer cells upon stimulation with IFNγ. 79 A specific STAT3 or AKT inhibitor could inhibit the IFNγ-induced PD-L1 expression in the same model. 79 Indeed, the activation of STAT3 by IFNγ, with subsequent increase in PD-L1 expression, may occur independent of the presence of an oncogenic alteration. 80 In KRAS-mutant cells, the MAPK pathway is implicated in the regulation of PD-L1 expression. 81 We believe that signaling pathways common in several types of tumors can differentially regulate immune responses, according to the driver oncogene, with a profound role of STAT3 in EGFR-mutant NSCLC (Figure 1).
Perspectives
STAT3 is an immune-response modulator. The role of STAT3 in innate resistance to EGFR TKIs
18
and the fact that EGFR or ALK inhibition decreases PD-L1 levels, provide the rationale for reconsidering immunotherapy strategies in these two subgroups of NSCLC patients. PD-L1 is high at baseline in oncogene-addicted NSCLC, but monotherapy with immune checkpoint inhibitors has not, to date, demonstrated encouraging results.17,82 Although a co-culture model did not show clear synergistic effect,
66
we believe that a combined approach with TKIs and anti-PD-1/PD-L1 compounds merits further investigation both in treatment-naïve and after development of acquired resistance to TKIs. Clinical trials are ongoing and phase I studies have shown activity and a good safety profile, with the exception of the combination of durvalumab with osimertinib.
9
Another strategy that merits to be investigated is the combination of STAT3 inhibitors with immune checkpoint blockade. Simple approaches, like repurposing drugs that affect the tumor microenvironment, can improve the efficacy of immune checkpoint blockade. For instance, pterostilbene, a natural methoxylated analogue of resveratrol, inhibits both STAT3 and Src and prevents the function of T-regulatory lymphocytes (Tregs).
83
Pentoxifylline, a drug used for chronic occlusive arterial disease, can also prevent the recruitment of Tregs through inhibition of c-Rel, a subunit of the nuclear factor kappa B (NF-
The relation of STAT3 with PD-L1 expression in oncogene-addicted tumors provides evidence that STAT3 can be predictive of response to immunotherapy, and should be further investigated. Given the IFNγ-mediated activation of STAT3, and the awareness that this axis is able to up-regulate PD-L1, our model including STAT3 assessment at baseline can indirectly help to define which patients have an active tumor-immune infiltrate to be awakened with anti-PD-1/PD-L1 treatment.
Footnotes
Acknowledgements
IA and NK performed the literature search and wrote the manuscript; all authors contributed to the review and approval of the final manuscript.
Funding
Work in Dr. Rosell laboratory is partially supported by a grant from La Caixa Foundation, and by a European Grant (ELBA no 765492).
Conflict of interest statement
The authors declare that there is no conflict of interest.