
Abstract
KRAS mutations represent one of the most prevalent oncogenic driver mutations in non-small cell lung cancer (NSCLC). For many years we have unsuccessfully addressed KRAS mutation as a unique disease. The recent widespread use of comprehensive genomic profiling has identified different subgroups with prognostic implications. Moreover, recent data recognizing the distinct biology and therapeutic vulnerabilities of different KRAS subgroups have allowed us to explore different treatment approaches. Small molecules that selectively inhibit KRAS G12C or use of immune checkpoint inhibitors based on co-mutation status are some examples which anticipate that personalized treatment for this challenging disease is finally on the horizon.
Introduction
Over the past decade, the development of genotype-directed therapeutic strategies has transformed the treatment options for patients with different types of cancer. In non-small cell lung cancer (NSCLC), somatic mutations in EGFR, BRAF and HER2 and rearrangements in ALK, ROS and RET have been validated as powerful predictive biomarkers, expanding treatment options for molecularly defined subsets of patients and highlighting the recognition that NSCLC represents a multitude of different malignancies. 1 However, no anti-RAS therapy has succeeded in the clinic in spite of being one of the most prevalent oncogenic driver mutations in these types of tumors. 2
RAS genes comprise the most frequently mutated gene family in human cancers and consists of three members: HRAS, KRAS and NRAS. These genes encode four closely related proteins (HRAS, KRAS4A, KRAS4B, NRAS). The RAS family encodes small enzymes that hydrolyze guanosine triphosphate (GTPases) that activate various signaling pathways such as RAF-MEK-ERK, PI3K-AKT-mTOR and RALGDS-RA. As a result, RAS genes have a crucial role in the regulation of cell proliferation, differentiation and survival. The mutated RAS isoform varies across type of tumors, with KRAS being the most common in lung, pancreatic and colon cancer, NRAS in melanoma and HRAS in bladder cancer. RAS mutations tend to be single base substitutions that lead to stabilization of GTP binding and causing the extracellular-signal-regulated kinase (ERK) signaling pathway to be active.
The mutational status of KRAS has been extensively studied in NSCLC by both conventional molecular techniques and next-generation sequencing3–5 (see Table 1 and Figure 1(a)). Most of the mutations involve exon 2 at codon 12 with G12C being the most frequent mutation followed by G12V, G12D, and G12A; the rest involve codon 13 and occasionally exon 3 at codon 61. KRAS mutations are present in approximately 30% of lung adenocarcinomas and 5% of squamous cell carcinomas. It is more common in western (26%) than in Asian (11%) populations and in smokers (30%) than nonsmokers (10%). 6 The smoking pattern has also been related to the type of KRAS mutation; transversion mutations (substitution of a purine nucleotide to a pyrimidine or vice versa) are more common in current or ex-smoker patients while never-smoker NSCLC patients have a higher frequency of transition mutations (purine to purine or pyrimidine to pyrimidine nucleotide changes).7,8
Descriptive of KRAS-mutated samples in the three datasets analyzed.
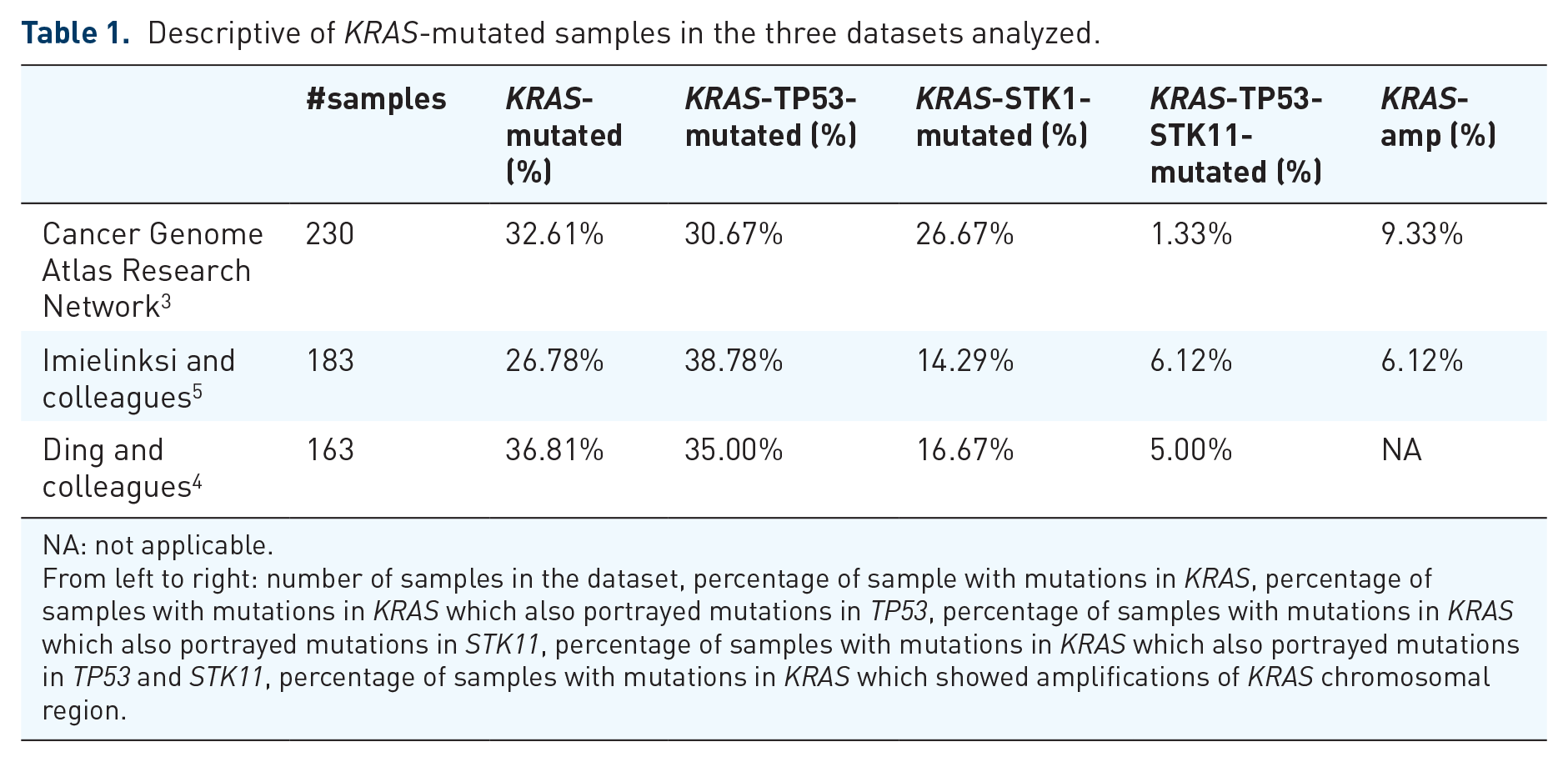
NA: not applicable.
From left to right: number of samples in the dataset, percentage of sample with mutations in KRAS, percentage of samples with mutations in KRAS which also portrayed mutations in TP53, percentage of samples with mutations in KRAS which also portrayed mutations in STK11, percentage of samples with mutations in KRAS which also portrayed mutations in TP53 and STK11, percentage of samples with mutations in KRAS which showed amplifications of KRAS chromosomal region.

It is well known that structural differences between various codon 12 and 13 mutations may affect GTPase activity and binding of effector proteins, like vascular endothelial growth factor/vascular permeability factor (VEGF/VPF).9–11 For example, GTPase activity of G12V RAS is less effective than activity of G12D Ras and binding of the GTP analogue GppNp to G12D Ras is weaker than its binding to G12V, which can be translated to an escape from the oncogenic GTP-bound state of G12D, whereas GTP tightly bound to G12V mutant Ras generates a more persistent, potentially oncogenic, signal. 9 In addition, mutations in codon 12 enhanced resistance to apoptosis, loss of contact inhibition, and predisposition to anchorage-independent growth, in contrast to mutations in codon 13, which increased sensitivity to apoptosis was associated with increased activation of the c-Jun-NH2-terminal kinase 1 pathway. 10
Finally, it has been suggested that different KRAS mutations can activate distinct signaling pathways. Thus, NSCLC cell lines with mutant KRAS G12A have activated phosphatidylinositol 3-kinase and mitogen-activated protein/extracellular signal-regulated kinase (MEK) signaling, whereas those with mutant KRAS G12C or mutant KRAS G12V have activated Ral signaling. 12 These different downstream effects may result in different prognostic significance and response to therapy among KRAS mutations. 13
Prognostic role of KRAS mutations in NSCLC tumors
Initially reported as a negative prognostic factor associated with early relapse and shortened survival, subsequent large randomized clinical trials with observational control arms have questioned this. The main reasons could be the use of detection methods with very different coverage, sensitivity and specificity, probably less comprehensive and accurate than the technologies in use nowadays (for example, targeted next-generation sequencing) and the recent recognition of heterogeneity within KRAS-driven NSCLC tumors.
The study E4592, a laboratory ancillary study on an Eastern Cooperative Oncology Group prospective randomized trial of postoperative adjuvant therapy (E3590), showed a 24% overall incidence of KRAS positive in 184 assessable tumors. Neither presence nor absence of p53 mutations, p53 protein expression or KRAS mutations correlated with overall survival (OS) or progression-free survival (PFS). 14 Similarly, the JBR.10 study, a North American intergroup trial in which patients were stratified according to nodal status and the presence or absence of a RAS mutation to randomly receive adjuvant cisplatin/vinorelbine or observation alone, demonstrated no significant prognostic value for RAS mutation in 450 patients, 117 of them with RAS mutations. 15 No differences in overall survival based on KRAS status were also found in the International Adjuvant Lung Cancer Trial study in which KRAS mutation was detected in 14% of patients with available samples 16 or in the Cancer And Leukemia Group B 9633 study, where the percentage of patients with KRAS mutation was 27%. 17
Similarly, several databases retrospectively analyzed the prognostic role of KRAS in the early stage setting. In particular, the European Early Lung Cancer project assessed the value of KRAS, TP53 and EGFR in 762 resected NSCLC patients with good quality frozen tissues from 12 centers in eight European countries. Mutation of none of the three genes appeared to carry a significant prognostic value, either as a whole or in specific histological subgroups. However, a separate analysis on the largest and most homogeneous subgroup revealed a borderline effect in patients carrying both TP53 and KRAS mutations [hazard ratio (HR) 3.26, 95% confidence interval (CI) 1.07–9.90; p = 0.038]. 18
On the other hand, several recent meta-analyses associated KRAS mutation status with worse OS, particularly in adenocarcinoma patients and an early setting.19–21
The heterogeneity of different KRAS mutation subtypes in terms of their prognostic value has been analyzed in several studies. Ihle and colleagues using integrated clinical data from a molecularly targeted clinical trial showed that patients whose tumors had either mutant KRAS G12C or G12V had worse PFS compared with patients whose tumors had other mutant KRAS proteins or wild-type KRAS. 12 Nadal and colleagues showed in 179 resected lung adenocarcinoma that patients with KRAS-G12C mutant tumors had significantly shorter disease-free survival (DFS) compared with tumors harboring other KRAS mutations or KRAS wild-type tumors. 22 In contrast, Izar and colleagues identified improved OS and DFS for G12C and G12V patients in 127 patients. 23 Similarly, Renaud and colleagues showed that KRAS G12V patients had significantly higher risk of recurrence, lower median OS and lower time to recurrence when compared with non-KRAS G12V, EGFR mutated or wild-type patients in 841 surgically treated French patients. 24 Finally, the largest report published with more than 1500 resected NSCLC patients (300 with mutations), showed that KRAS mutation status had absolutely no prognostic effect on OS in the whole group or when analyzed by the adenocarcinoma subset (accounting for almost 70% of mutations), the KRAS subtypes (codon 12 or 13 mutations) or even by the different codon 12 subgroups in the observational arm. 25
Apart from the early setting, the prognostic value of KRAS mutation has also been explored in advanced disease. KRAS mutation status has demonstrated not to be an independent prognostic factor in stage IV patients treated with conventional therapy 26 or EGFR tyrosine kinase inhibitors (TKIs) 27 although, once again, the differential role of distinct codon 12 subtypes has been highlighted. Renaud and colleagues analyzed the ability of specific mutations to predict site-specific recurrence and metastases. In their series, comprising 481 patients who experienced thoracic or extra-thoracic recurrence and metastases after surgery, KRAS G12C and G12V were predictive of bone and pleuro-pericardial metastasis, respectively, while KRAS G12V was inversely associated with lung recurrence. 28
Predictive role of KRAS mutations in NSCLC tumors
Regarding the predictive role, there was no significant effect of KRAS mutations on benefit from adjuvant chemotherapy with respect to OS or DFS in the LACE-Bio project. However, the small subset of 24 patients with codon 13 mutations had significantly poorer outcomes with chemotherapy than observation alone. 25 In the advanced setting, the predictive value also remains unclear. This is probably due to the fact that most of the studies comprise retrospective small sample sizes series, mixed different NSCLC histologies and diverse therapeutic settings. 29
Garassino and colleagues reported that the G12C variant was associated with a reduced response to cisplatin and an increased sensitivity to paclitaxel and pemetrexed in cell lines. Systematic analysis in cell lines of drug uptake, DNA adduct formation and DNA damage responses implicated in cisplatin adducts removal, revealed that G12C mutation might be particular because it stimulates base excision repair to rapidly remove platinum from DNA even before the formation of cross-links. 30 Conversely, the G12V mutant yielded a strong sensitivity to cisplatin compared with wild-type clones and a slight resistance to treatment with pemetrexed. 31 The expression of G12D mutants resulted in resistance to paclitaxel treatment and sensitivity to sorafenib.
The predictive value of KRAS mutations has also been addressed in patients receiving EGFR TKIs. A small retrospective study reported no differences in terms of response to EGFR TKIs in 67 EGFR wild-type NSCLC patients when comparing among codon 12 and codon 13-mutated KRAS tumors. However, codon 13-mutated patients experienced a significantly shorter PFS and OS compared with codon 12-mutated patients and KRAS wild-type group. 32 In a pooled analysis of four trials of EGFR TKIs versus placebo, RAS status was known for 1362 of 2624 patients (785 receiving EGFR TKIs and 577 receiving placebo) and 275 harbored KRAS mutations (248 at codon 12, 15 at codon 13, 12 at other codons). KRAS mutations were neither prognostic nor predictive of benefit from EGFR TKIs; however, the pooled analysis demonstrated a potential OS benefit for EGFR TKIs in patients with KRAS G12D/G12S transition and a trend toward inferior survival with EGFR TKI for patients with G12C/G12V transversion that became significant in the adenocarcinoma subpopulation, particularly in G12V patients. According to the authors, these observations of better prognosis for G12C/G12V yet poorer treatment effect from EGFR TKIs and poorer prognosis for G12D/G12S and apparent benefit from EGFR TKIs are intriguing and require prospective validation. 27
The relationship between KRAS status and response to radiotherapy has been analyzed in several studies. In vitro and clinical studies suggest that KRAS also predicts for poorer response to radiation although this phenomenon is still understudied. Tang and colleagues have characterized the effects of suppressing focal adhesion kinase (FAK). With functional experiments, they found that, in mutant KRAS NSCLC cells, FAK inhibition resulted in persistent DNA damage and susceptibility to exposure to radiotherapy. 33 Wang and colleagues recently describe a radiation resistance phenotype conferred by a stem-like subpopulation characterized by mitosis-like condensed chromatin, high CD133 expression, invasive potential, and tumor-initiating properties. This subset of KRAS-mutated lung cancers is enriched for co-occurring genomic alterations in TP53 and CDKN2A that could have implications for prognostic and therapeutic strategies. 34 Finally, differences according to amino acid substitutions have also been related to radiation, being G12V or G12C status associated with both poor response rate (RR) and OS in NSCLC patients with brain metastasis. 35
Targeting KRAS in NSCLC patients
In the era of personalized treatment, RAS still seems an elusive target. 36 In 2013, the US National Cancer Institute launched the RAS Initiative. Since then, novel therapeutic approaches to hit KRAS-mutated NSCLC have been developed and numerous clinical trials are currently ongoing.
The feasibility of a direct targeting of KRAS has been extensively explored and different small molecules have been analyzed. However, their crucial role in normal physiology became a challenge and substantial toxicity appeared as a result of the indiscriminate inhibition of both wild-type and mutant KRAS. In this regard, recent studies have focused on mutant-specific inhibition.37–39 In particular, results from ARS-853, a small molecule with potent antitumor activity against G12C KRAS, are largely awaited. 40
However, it is important to remark that probably not all KRAS-mutated lung cancers will show the same degree of ‘KRAS addiction’ In this sense, it has been demonstrated in lung and pancreatic cancer cells that two classes of KRAS-mutated tumors existed, those that requires KRAS activation to maintain viability and those that do not. Comparing these two classes of cancer cells revealed a gene expression signature in KRAS-dependent cells, associated with a well-differentiated epithelial phenotype, which was also seen in primary tumors. Several of these genes encode pharmacologically tractable proteins, such as Syk and Ron kinases and integrin beta6, depletion of which induces epithelial-mesenchymal transformation (EMT) and apoptosis specifically in K-Ras-dependent cells. 41 These findings suggested that EMT regulators in ‘KRAS-addicted’ cancers represent candidate therapeutic targets, and that direct targeting of KRAS in ‘KRAS-independent’ tumors will provide little therapeutic benefit.
Another area of great interest when targeting KRAS has turned to downstream effectors, in particular the inhibition of MEK1 and MEK2 signaling. Further downstream of MEK has also been explored and selective inhibitors of CDK4 and CDK6 are currently being assessed. 42
Trametinib, a reversible MEK1/2 inhibitor, was compared with docetaxel in a randomized phase II study enrolling pretreated KRAS-mutant NSCLC patients. Regrettably, PFS and overall response rate (ORR) were similar in both arms. 43 Trametinib has also been tested in combination with chemotherapy, showing interesting outcomes in an exploratory subpopulation analysis of a phase I study in the subset of patients with G12C KRAS mutations (4 of 10 patients with confirmed response and 8 of 10 with disease control). 44
Selumetinib is another orally selective potent inhibitor of MEK1/MEK2 kinases. The combination of docetaxel and selumetinib showed promising results in the second-line setting for the subset of NSCLC patients with KRAS mutation. In fact, in the SELECT study, a randomized phase II trial, the combination of docetaxel plus selumetinib achieved a statistically significant increase in median PFS, ORR and a promising trend for superior OS in comparison with docetaxel alone. 45 However, the results were not confirmed in the phase III trial comparing selumetinib plus docetaxel with placebo plus docetaxel. 46
In trying to identify subsets of patients suitable to receive selumetinib plus docetaxel, a retrospective analysis of the SELECT study was carried out. 47 The results showed superior PFS and OS in patients with KRAS G12C or G12V mutations that received selumetinib plus docetaxel but the trend was no statistically significant. The secondary analysis of the Select-1 phase III study also obtained similar results. They explored the possibility that KRAS G12C or G12V mutations had greater sensitivity to selumetinib. However, no treatment effect on PFS was found based on different KRAS mutation subtypes determined by next generation sequencing (NGS). Finally, the influence of PD-L1 status to identify subsets of patients to benefit from selumetinib/docetaxel was also explored but, once again, there was not significant impact on PFS, OS or ORR in either treatment arm. 48
Analyzing the role of microRNAs (miRNAs) in the regulation of EGFR signaling pathways may provide insights into improving the management of KRAS-mutant lung cancers. Recently, miR-29b has been proposed as an important target for upregulation by mutant KRAS in these tumors. In KRAS (G12V)-transduced bronchial epithelial (BEAS-2B) cells, introduction of anti-miR-29b constructs increased the sensitivity to apoptosis by targeting TNFAIP3/A20, a negative regulator of nuclear factor (NF)-κB signaling. Accordingly, overexpression of a miR-29b-refractory isoform of TNFAIP3 restored NF-κB and extrinsic apoptosis, confirming that TNFAIP3 is a functionally relevant target of miR-29b. 49
Overexpression of miR-31 has been observed in pancreatic and colorectal cancer harboring mutations in KRAS. 50 In addition, miR-31 has been proposed as a driver of lung tumorigenesis that promotes mutant KRAS-mediated oncogenesis by directly targeting and reducing expression of negative regulators of RAS/MAPK signaling. 51 On the other hand, there are miRNAS that are downregulated by RAS. Thus, Ras activation leads to repression of the miR-143/145 cluster in human tumor cell lines, and loss of miR-143/145 expression is observed frequently in KRAS-mutant pancreatic cancers. miR-143/145 down-regulation requires the Ras-responsive element-binding protein (RREB1), which represses the miR-143/145 promoter. Additionally, KRAS and RREB1 are targets of miR-143/miR-145, revealing a feed-forward mechanism that potentiates Ras signaling. 52 All these data suggest that some micro-RNAs might be very interesting therapeutic targets in KRAS-mutated lung cancer.
Very recently another area of interest focused on the biological relevance of co-mutations in KRAS-mutant NSCLC patients has emerged (see Table 1 and Figure 1(b)). Skoulidis and colleagues demonstrated that genetic alterations in STK11/LKB1, TP53, and CDKN2A/B define three major subgroups of KRAS-mutant lung adenocarcinoma. 53 They reported that LKB1-deficient tumors exhibit a ‘cold’ tumor immune microenvironment with reduced expression of several immune checkpoint effector/mediator molecules while those tumors with TP53 alterations demonstrated higher levels of inflammatory markers and evidence of active immunoediting. Similarly, in a large cohort of 442 resected tumors, TP53 was strongly associated with enhanced proliferation and STK11 with suppression of immune surveillance. 54
STK11/LKB1 is a gene commonly lost in NSCLC KRAS-mutated tumors. LKB1 mutations are associated with reduced expression of PD-L1 in mouse, patient tumors and tumor-derived cell lines while genetic ablation of LKB1 results in accumulation of neutrophils with T-cell-suppressive effects. 55 Calles and colleagues retrospectively analyzed 154 KRAS-mutant NSCLC tumors for LKB1 using immunohistochemistry. LKB1 expression was lost in 30% of KRAS-mutant NSCLC (smokers 35% versus never-smokers 13%) and was associated with a more aggressive clinical phenotype. LKB1 expression did not correlate with a specific KRAS mutation but was more frequent in tumors with KRAS transversion mutations. 56 Preclinical studies using genetically engineered mouse model (GEMM) also achieved similar results. Chen and colleagues showed that selumetinib/docetaxel was more effective than docetaxel alone in a KRAS G12D GEMM but not in a KRAS G12D that showed LKB1 loss. 57
The relevance of these findings is related to the potential predictive value of the different co-mutations status in guiding anti‑PD-1/PD-L1 immunotherapy in KRAS-mutated NSCLC. Skoulidis and colleagues analyzed the clinical outcomes of different KRAS subgroups in 35 patients with metastatic disease treated with immune checkpoint inhibitors. They did not find any impact of different KRAS alleles (G12C/G12V/G12D) on PFS or ORR. However, KRAS subgroups with different co-mutation exhibited significantly different outcomes. In particular, mutations in KRAS and STK11/LKB1 were associated with an inert tumor immune microenvironment and poor clinical response to immune checkpoint blockade. 58 Similarly, Dong and colleagues showed that TP53/KRAS co-mutated subgroup manifested exclusive increased expression of programmed death-ligand 1 (PD-L1), a highest proportion of PD-L1+/CD8A+ expression and a remarkable clinical benefit to PD-1 inhibitors. 59 Lastly, a recent retrospective study analyzed the clinical and molecular characteristics of 114 KRAS-mutant NSCLC patients and the correlation with the immunohistochemistry (IHC) expression of PD-1, PD-L1, and PD-L2. They found that PD-L1 and PD-L2 were more frequently expressed when LKB1 was intact. However, the difference was statistically significant only for PD-L2 cases, probably because of the limited number of cases with PD-L1-positive expression for comparison. 60
In summary, KRAS mutations represent one of the most prevalent oncogenic driver mutations in NSCLC. For many years we have unsuccessfully addressed KRAS mutation as a unique disease. The recent widespread use of comprehensive genomic profiling has identified different subgroups with prognostic implications. Moreover, recent data recognizing the distinct biology and therapeutic vulnerabilities of different KRAS subgroups have allowed us to explore different treatment approaches. Small molecules that selectively inhibit KRAS G12C or use of immune checkpoint inhibitors based on co-mutations status are some examples which anticipate that personalized treatment for this challenging disease is finally on the horizon.
Footnotes
Funding
This work was supported by the ISCIII: PIE15/00050, FEDER Centro de Investigaciones Biomédicas en Red en Cáncer (CIBER-ONC), Madrid, Spain.
Conflict of interest statement
The authors declare that there is no conflict of interest.