
Abstract
Over the past decade there have been significant advances in the molecular characterization of colorectal cancer (CRC) that are driving treatment decisions. Expanded RAS testing beyond KRAS exon 2 was established as crucial for identifying patients who will respond to anti-epidermal growth factor receptor (EGFR) therapies and low-frequency mutations in RAS/tumor heterogeneity are gaining recognition as potential mechanisms of resistance. Despite this progress, the fact that we do not understand why left-sided but not right-sided tumors have improved outcomes following anti-EGFR therapy highlights our superficial understanding of this disease. Even with few new targeted agents receiving approval in CRC, the incorporation of next-generation sequencing into clinical decision making represents an important step forward. Biomarkers such as BRAF mutations, microsatellite instability, and HER2 amplification represent promising molecular aberrations with therapies in various stages of development, and highlight the importance of companion diagnostics in supporting targeted agents. In this review, we will discuss the importance of incorporating biomarkers into clinical decision making and regimen selection in CRC. We will particularly focus on the recent evidence suggesting an important role for tumor location in selecting first-line therapy, the importance of recent advances in biomarker development and molecular subtyping, as well as recently approved agents (regorafenib and TAS-102) and promising targeted agents that have the potential to change the standard of care.
Introduction
Colorectal cancer (CRC) is the third leading cause of cancer and cancer-related death. 1 Approximately 20% of patients present with de novo metastatic disease, and 25–30% of patients with stage II/III disease will have a recurrence within 5 years of a curative intent surgery. 2 Surgical resection and locoregional ablative therapies can result in cures for carefully selected patients with oligometastatic disease, however most patients with disseminated disease have a condition that is not curable and will require systemic therapy. First- and second-line therapies typically consist of a fluoropyrimidine doublet (FOLFOX/CAPOX or FOLFIRI/CAPIRI) combined with a biologic targeting either angiogenesis (bevacizumab, ramicurumab, ziv-aflibercept) or the epidermal growth factor receptor (EGFR) (cetuximab or panitumumab) in patients with RAS wild-type tumors.3,4 In some patients, sequential single-agent therapy is a reasonable treatment approach that does not appear to be considerably less effective than combination therapy. 5 Maintenance chemotherapy with a fluoropyrimidine with or without bevacizumab is an option for carefully selected patients whose disease has responded to chemotherapy as a way of providing a treatment break and appears to result in better outcomes than complete chemotherapy-free intervals.6,7 Third-line options for patients with RAS wild-type disease that has not previously been treated with anti-EGFR therapy include panitumumab or cetuximab with or without cytotoxic chemotherapy.8,9 For patents with disease that has previously progressed on anti-EGFR agents or who have RAS mutant disease, regorafenib and TAS-102 may be used.10,11
While there have been relatively few agents with novel mechanisms introduced into the treatment algorithm for metastatic CRC (mCRC) over the past decade, there has been considerable advancement in the molecular characterization of mCRC. We now understand the importance of RAS and BRAF mutations as predictive and prognostic markers and are beginning to understand that CRC is made up of distinct molecular subtypes that are each driven by unique biologic aberrations. 12 Most recently, the disparate response of right- and left-sided primary tumors to anti-EGFR therapy has underscored the importance of subgrouping mCRC. Accompanying the appreciation that mCRC needs to be subgrouped has been the growing ability to use this information clinically. Significant advancements in tissue-sequencing platforms and the advent of liquid biopsies are allowing molecular characterization to guide therapy and is improving our ability to understand genetic evolution and tumor heterogeneity. 13 In this review, we will discuss the recent progress in sequencing agents to improve outcomes, novel agents that have or are on the verge of changing practice, and the importance of using companion biomarkers and molecular subtyping to guide therapeutic decisions. A potential treatment algorithm incorporating this information is highlighted in Figure 1.

Potential treatment options for patients with metastatic colorectal cancer that incorporates molecular characteristics and anatomic site into the decision-making process.
Sequencing of agents in first-line and second-line therapy and tumor sidedness
In patients receiving a fluoropyrimidine doublet for first- or second-line therapy, the order of oxaliplatin and irinotecan components has not been shown to impact outcomes and the decision is often based on regional practice patterns, toxicity profiles, and patient comorbidities.14,15 There have been several failed attempts to identify biomarkers to help in selecting the optimal first-line cytotoxic backbone. 16 Patients who require rapid tumor shrinkage (i.e. those who may be candidates for metastectomy) or those with negative prognostic features, such as BRAF mutations, may benefit from the use of the cytotoxic triplet FOLFOXIRI plus bevacizumab. 17 For patients with RAS mutant disease, doublet or triplet therapy with bevacizumab is the standard first-line option.
Recent evidence suggests that primary tumor location may not only be prognostic, but may also have a predictive role in RAS wild-type mCRC. Left-sided tumors have been shown to have a better prognosis compared with right-sided tumors. 18 This is likely in part due to a number of molecular features that are more common in right-sided tumors, such as BRAF mutations and microsatellite instability (MSI). 19 More comprehensive gene expression based subtyping has demonstrated that right-sided tumors are more commonly associated with an immunologically active consensus molecular subtype (CMS-I), characterized by higher rates of MSI, CpG island methylator phenotype (CIMP-H), hypermutation, immune infiltration and activation, and worse survival following relapse. 12
Highlighting the differential molecular pathways affected in mCRC have been recent retrospective analyses comparing outcomes following anti-EGFR therapy. Figures 2 and 3 summarize the findings of these studies, all of which have defined right-sided tumors as being those from the cecum to the transverse colon (except for 80405 which omitted transverse tumors from the analysis). Taken together, these results suggest that having a left-sided primary tumor is predictive of improved median overall survival (mOS) and median progression-free survival (mPFS) following anti-EGFR therapy in the first-line setting for patients with RAS wild-type disease.20–22 This effect appears to be independent of the higher rate of BRAF mutations in right-sided tumors. In contrast, a trend towards worsened outcomes for right-sided tumors when treated with anti-EGFR agents was noted. CRYSTAL, FIRE-3, PRIME, PEAK, 80405, 181 and NCIC CO.17 have all shown a similar direction of effect, while CRYSTAL, FIRE-3 and 80405 were analyzed with tests of interaction confirming an interaction between side and treatment response (p < 0.05). 23 This differential response was also shown in the third-line setting in NCIC CO.17, in which left-sided tumors had improved outcomes following cetuximab compared with best supportive care but right-sided tumors did not.
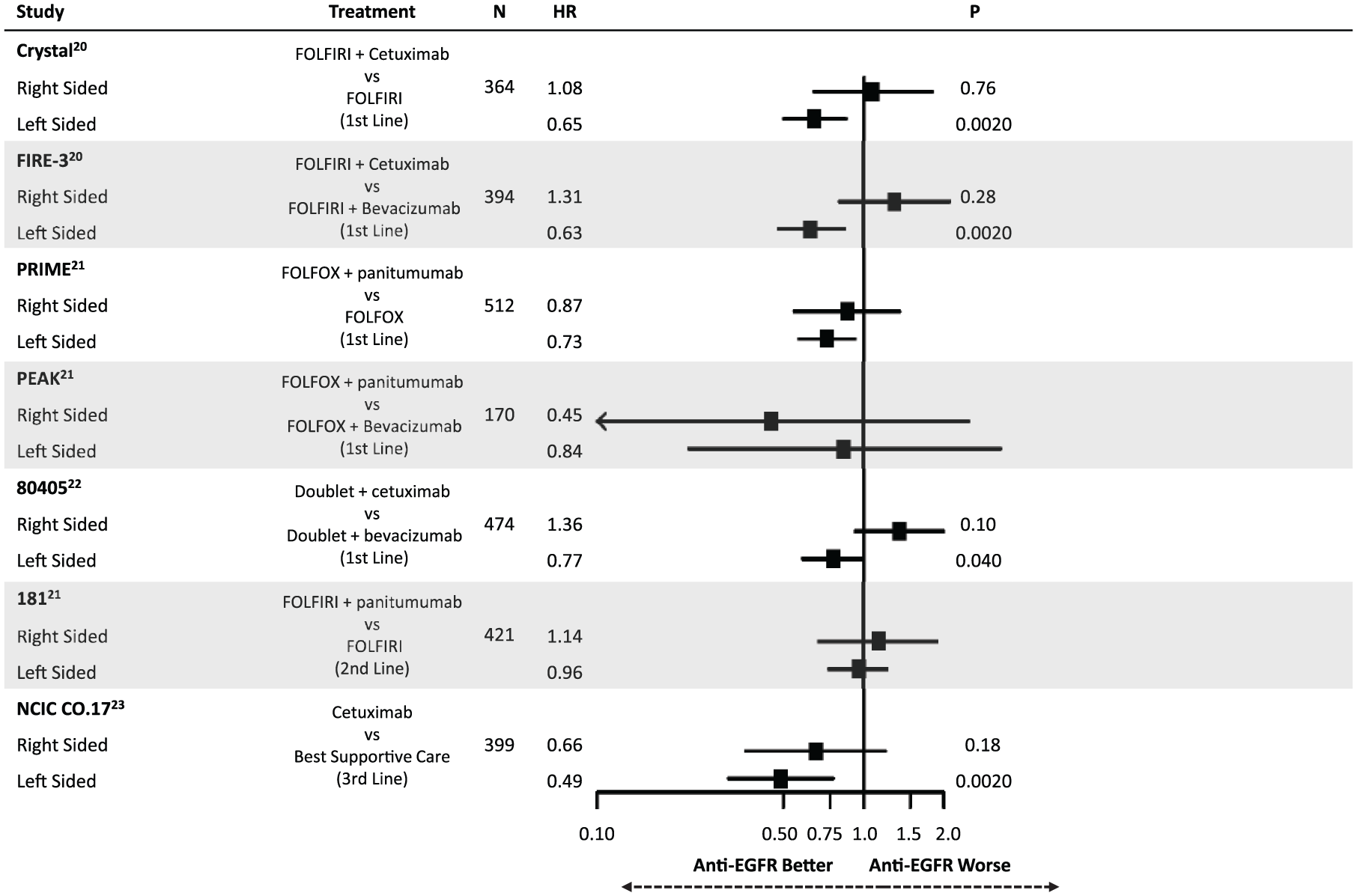
Summary of evidence demonstrating a differential impact of anti-epidermal growth factor receptor (EGFR) therapy on overall survival based on primary tumor location.
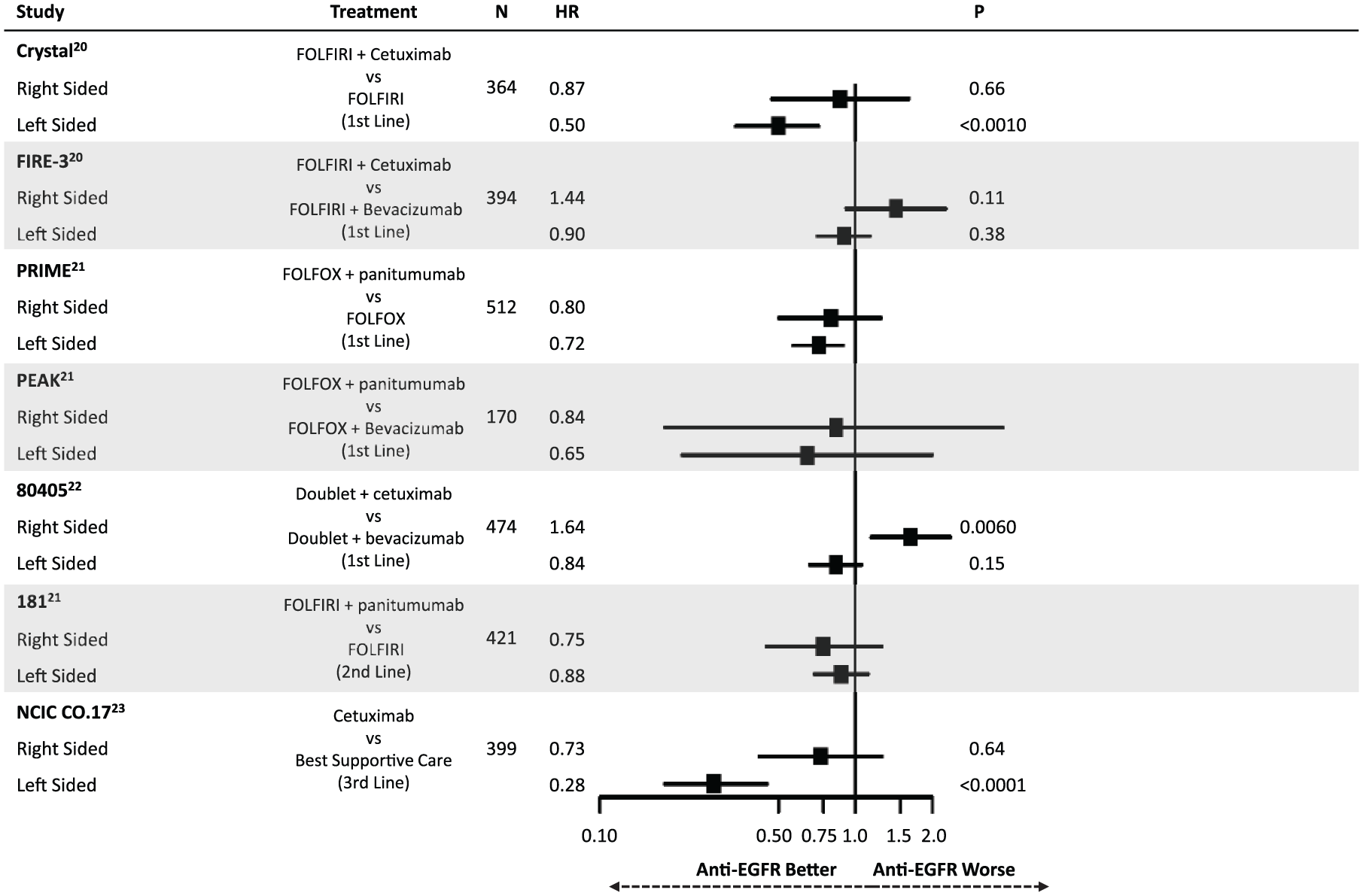
Summary of evidence demonstrating a differential impact of anti-epidermal growth factor receptor (EGFR) therapy on progression-free survival based on primary tumor location.
Though the data supporting tumor location as a predictive marker are based on retrospective analysis of randomized trials, the consistency of these findings strongly supports the use of sidedness in clinical practice when selecting therapy. Importantly, these findings were not only statistically significant, but were clinically meaningful. For example, in 80405 mOS for left-sided patients treated with a doublet plus cetuximab was 39.3 months compared with 32.7 months when treated with a doublet plus bevacizumab. More shocking was the difference for right-sided tumors. When receiving a doublet plus cetuximab, mOS was 13.7 months compared with 29.2 months for right-sided patients receiving a doublet plus bevacizumab. 22 While the molecular underpinnings responsible for this differential response is unclear, it highlights the importance of better defining the biology underlying these distinct clinical behaviors.
Molecular subtyping of CRC
Advancements in next-generation sequencing (NGS) have allowed more comprehensive genotyping of tumors with assays of higher sensitivity. This has allowed us to identify that there are multiple distinct but convergent pathways that lead to treatment resistance in mCRC. For example, following anti-EGFR therapy numerous different alterations have been demonstrated to result in resistance, including acquired RAS mutations, EGFR mutations, ERBB2 amplifications, and MET amplifications. 24 As we gain a better understanding of how and in which patients these resistance mechanisms emerge, we may be able to use combination therapies to prolong treatment responses.
One of the major diagnostic changes that can be addressed by NGS has been the move from single gene assays to multiplexed panels. These panels allow multiple samples from different patients to be tested concurrently for mutations in multiple genes. This facilitates screening for molecularly targeted trials and helps reduce the risk of tissue exhaustion during pathologic workup. 25 As the cost of NGS continues to fall, a panel approach may provide genotyping of a large number of genes for comparable cost to a single gene polymerase chain reaction (PCR) test. 26
Expanded RAS testing with increased sensitivity identifies a better population for anti-EGFR treatment
The increased breadth of NGS assays is particularly important for RAS testing. While KRAS exon 2 mutations were initially shown to result in resistance to anti-EGFR therapy, repeat analysis of the PRIME trial demonstrated that mutations in exon 2, 3, and 4 of both KRAS and NRAS provide resistance to EGFR-directed therapy.8,27 With expanded RAS testing, mutations are present in around 56% of mCRCs, while previous PCR-based assays only identified mutations in 40–45% of patients. 28 The importance of detecting these mutations is that these patients may not only lack benefit, they may be harmed by receiving cetuximab/panitumumab. Besides added toxicity, there appears to be a signal towards harm in patients who are RAS mutant receiving anti-EGFR therapy. 29
Retrospective analysis of nonmicrodissected tumors from several trials has shown that low-frequency RAS mutations that would not be identified by standard testing may also provide resistance to anti-EGFR therapy. While standard clinical assays previously defined patients as having RAS mutation when a variant allele fraction was over 10%, rates as low as 0.1% have been shown to confer resistance, although allele frequency cutoffs have not been rigorously defined. In the CAPRI-GOIM trial, the use of a high-sensitivity assay testing for the same mutations as a previously performed clinical assay was able to identify an additional 15.9% of patients as having KRAS mutation. 29 Multiple groups have reported resistance from low-frequency RAS mutations, however in the CRYSTAL trial, the impact of mutant RAS allele fraction appeared to follow a gradient. Patients harboring mutations at extremely low frequencies still received some benefit from adding cetuximab to chemotherapy. 30 It is not clear whether these low-frequency mutations represent subclonal populations and tumor heterogeneity or clonal tumors missed with standard assays due to lack of microdissection of tumor from contaminating stroma.
Liquid biopsies
Liquid biopsies assessing cell-free DNA (cfDNA) present a novel approach to obtain real-time assessments of the most prevalent genotypic clones present in a tumor. Using only a few milliliters of blood, NGS panels can identify biomarkers to guide therapy, such as RAS status, and may also be used to identify resistance mechanisms that emerge during treatment, potentially before radiographic progression.31,32 These panels allow the tracking of clonal dynamics during therapy and provide clinicians with more relevant mutation screening than relying on results from archival tissue. In addition, cfDNA may allow us to reuse systemic therapies as resistant clonal populations wax and wane. This strategy has been previously employed to reuse anti-EGFR therapy in mCRC and allowed retreatment with second responses after disappearance of resistant clones. 31
Consensus molecular subtypes of CRC
While single gene alterations allow us to test targeted agents in select groups of patients, mCRC can also be grouped into CMS based on gene expression profiles. 12 While some mutations are more common in certain CMS, particular driver mutations appear to be less important than biological signatures associated with each category. Using this classification, patients can be grouped into MSI/immune (CMS1), canonical/WNT (CMS2), metabolic (CMS3), and mesenchymal (CMS4) profiles. Though these subtypes are still not ready for introduction into standard clinical practice, they present an opportunity to guide targeted therapies with underlying biologic rationale and will hopefully help to improve the success of targeted agents in mCRC trials.
Distinct molecular aberrations and experimental agents
BRAF-mutated mCRC
BRAF mutations occur in 8–10% of mCRCs, frequently co-occur with MSI, and usually occur in patients who are RAS wild type.32,33 These mutations are associated with poor prognosis and may also predict reduced response to anti-EGFR therapy.34–36As BRAF is downstream of RAS it intuitively follows that BRAF mutations may result in resistance to anti-EGFR therapy. Two large meta-analyses both suggested the addition of anti-EGFR therapy to standard chemotherapy did not result in improved mPFS or mOS in patients who were RAS wild type and BRAF mutant.37,38 In-vitro experiments have suggested that patients with BRAF mutation may have sensitivity to microtubule inhibitors such as vinolrelbine or vinblastine, however this has not been validated in prospective trials. 39
The use of upfront FOLFOXIRI plus bevacizumab for BRAF-mutated mCRC is an option based on retrospective studies and a single-arm prospective trial. In TRIBE, treatment with FOLFOXIRI plus bevacizumab was associated with a trend towards improved OS [hazard ratio (HR) 0.54, 95% confidence interval (CI) 0.24–1.20] and PFS (HR 0.57, 95% CI 0.27–1.23) compared with FOLFIRI plus bevacizumab among the 28 patients with BRAF mutation. 17 Loupakis and colleagues performed the only prospective study of FOLFOXIRI plus bevacizumab for BRAF-mutant mCRC in a single-arm phase II trial after noting impressive results in a retrospective subgroup analysis of 10 patients with BRAF mutation from a molecularly unselected phase II trial. 40 In their prospective trial of FOLFOXIRI plus bevacizumab in 15 patients with BRAF mutation, they reported an impressive mOS (24.1 months) and mPFS (9.2 months). 41
The evidence for a cytotoxic triplet in this group is weak and further research is needed to deliver successful treatment options for these patients. Evaluation of BRAF inhibition in mCRC has produced much less impressive results than in melanoma. Single-agent BRAF inhibition resulted in a 5% response rate, while dual BRAF plus MEK inhibition is similarly not substantially active.42,43 To date, the highest response for targeted agents in BRAF-mutated mCRC has been 35% with the combination of vemurafenib, cetuximab, and irinotecan. 44 A phase II study using this regimen demonstrated an improvement in PFS from 2.0 to 4.4 months (p < 0.001) with the addition of vemurafenib to cetuximab and irinotecan. 45 Numerous other trials are currently underway evaluating the role of combining BRAF, MEK, and EGFR inhibitors.
In patients with MSI-H BRAF-mutant CRC, there is a lack of evidence to guide whether selecting immunotherapy or a BRAF inhibitor in combination with other agents would represent a better approach. Given the slightly higher response rates seen in CHECKMATE 142 (25.5% with nivolumab alone versus 33.3% for nivolumab + ipilimumab) compared with SWOG S1406 (16% with vemurafenib, cetuximab, and irinotecan) and the potential for prolonged periods of disease control in those patients who do respond to immunotherapy, we would favor an immunotherapeutic strategy in patients with BRAF-mutant MSI-H. However, this is not founded on strong evidence.45,46 As will be discussed later, mitogen-activated protein kinase (MAPK) modulation may also play a role in modulating the immune response. There may be benefit to targeting both pathways concurrently, however this has not yet been evaluated clinically.
Microsatellite instability and immune checkpoint inhibitors
Deficient DNA mismatch repair results in genomic instability and leads to expansion of repetitive elements throughout the genome called MSI. MSI is present in approximately 15% of all CRCs, with 4% occurring due to inheritance of germline mutations in MLH1, MSH2, MSH6, PMS2, or EPCAM as part of ‘Lynch syndrome’. The remaining cases are due to somatic alterations, most commonly resulting from hypermethylation of the MLH1 promoter. 47 In the metastatic setting, the incidence of MSI is closer to 5%.48,49 Testing for MSI status has previously shown utility in identifying patients with stage II disease who will not benefit from adjuvant chemotherapy. 50 In the metastatic setting, MSI is useful to identify incident cases of Lynch syndrome so that family members can undergo screening. An effective approach to screen for Lynch syndrome is to perform immunohistochemistry staining for DNA mismatch repair proteins and if one of these proteins has absent staining, to perform BRAF mutation analysis. BRAF mutations commonly occur in MSI tumors, however they almost exclusively occur in patients with sporadic MSI due to MLH1 promoter methylation and Lynch syndrome can effectively be ruled out.51–53 Figure 4 highlights a Lynch screening diagnostic algorithm based on the Evaluation of Genomic Applications in Practice and Prevention Working Group 2009 guideline. 54

Microsatellite instability testing algorithm. Either immunohistochemistry or microsatellite instability (MSI) polymerase chain reaction based assessment are acceptable, however immunohistochemistry is more cost effective. Each test will miss about 5–15% of Lynch syndrome cases and an alternate test should be considered in cases with a high pretest probability if results are negative. Consider a genetics referral for any high-risk family history with negative testing.
MSI ascertainment is also useful in mCRC to identify patients who are candidates for immune checkpoint inhibitors. In Le and colleagues’ phase II study of pembrolizumab in mCRC there were no responses reported in microsatellite stable (MSS) tumors, while there was a 40% response rate in patients with MSI. 55 Similarly, patients with MSI treated with the combination of nivolumab plus ipilimumab had response rates of 33.3% compared with 5% in MSS tumors. 46 This differential is hypothesized to be due to the elevated mutation burden in MSI which subsequently results in neoepitopes that may be targets for an activated immune system.56,57 In addition, an elevated neoantigen load has been associated with increased tumor-infiltrating lymphocytes in both MSI and MSS mCRCs, suggesting a potential mechanism for increased response. 58 While the optimal use of immunotherapy and potential combinatorial strategies that may improve efficacy are still under investigation, MSI testing should be offered to patients eligible for an immune checkpoint inhibitor and these agents are recommended in the National Comprehensive Cancer Network (NCCN) guidelines as second- or third-line therapy for MSI-H tumors. 4
For patients with MSS tumors, immunomodulatory strategies to turn these ‘cold’ tumors into ‘hot’ immunogenic tumors are also currently under investigation. Recently, Bendell and colleagues presented a phase Ib study looking at the combination of cobimetinib and atezolizumab in non-MSI-H tumors. 59 The rationale for this approach stems from the fact that preclinical models in both melanoma and CRC demonstrate that MEK inhibition can result in increased tumor-infiltrating lymphocytes, upregulation of programmed death-ligand 1 (PD-L1) and may improve responses to immunotherapy.60,61 This immune modulation is felt to be due to the role of MAPK signaling in antigen presentation and T-cell receptor signaling. 62 In Bendell and colleagues’ study, an objective response rate of 20% was noted. Though this may be promising, 70% of the patients in their study had undocumented MSI status, leaving interpretation of these results in the context of MSS tumors challenging.
ERBB2 amplifications
ERBB2 amplifications are present in 3–4% of mCRCs and mutations occur in 1–2% of patients.63,64 Amplifications result in lower response rates and shorter PFS during anti-EGFR therapy in the metastatic setting, however the functional impact of mutations is unclear.65,66 In patients with stage III disease, patients with ERBB2 amplifications or mutations had a shorter time to recurrence (HR 1.55, 95% CI 1.02–2.36, p = 0.04) and overall survival (HR 1.57, 95% CI 0.99–2.5, p = 0.05). 67 Despite the negative implications of these alterations, they may provide a novel target for systemic therapy. In the HERACLES trial, trastuzumab and lapatinib resulted in response rates of 30% in heavily pretreated patients with ERBB2 amplification. 68 Similarly, trastuzumab and pertuzumab demonstrated response rates of 23% and disease control rates of 69% in the colorectal arm of a basket study. 69 Testing for ERBB2 amplifications has not been standardized, however an algorithm similar to breast cancer seems appropriate, with immunohistochemistry (IHC) performed initially, followed by confirmatory in situ hybridization for all scores of 2+ and 3+.70,71 As NGS panels become more widely used, the application of panels to call copy number information is likely to play an important role in screening for this rare subtype.
Fusion proteins
A number of recurring fusion proteins resulting from chromosomal translocations have been described in CRC and represent potential targets for drug development. R-spondin fusions appear to be the most common, present in up to 10% of patients. They are mutually exclusive with APC mutations and result in aberrant Wnt signaling. 72 Targeting R-spondin with either Porcupine (PORCN) inhibitors to block Wnt secretion or monoclonal antibodies to the fusion protein has resulted in impressive responses in patient-derived xenograft models, however clinical activity has not yet been demonstrated.73,74 ALK and RET fusion proteins are both present in less than 1% of patients. Similar to ALK fusion positive lung cancers, colorectal tumors with ALK translocations appear sensitive to crizotinib and entrectinib in preclinical work.75,76 RET fusion proteins have been associated with clinical responses to regorafenib, a multikinase inhibitor which targets RET, and in vitro studies support the fact that tumor cells with this alteration may be sensitive to RET-targeted agents. 77
Novel agents
Vascular endothelial growth factor inhibition beyond first line
The use of bevacizumab in combination with first-line therapy was first demonstrated to improve survival when combined with irinotecan, fluorouracil, and leucovorin (IFL), when it improved mOS by nearly 5 months. 78 Recent estimates of impact have shown a more tempered benefit. 79 After progressing on first-line therapy, several trials evaluating bevacizumab beyond progression have shown small improvements in mOS.80,81 Alfibercept has been evaluated in combination with FOLFIRI in patients previously treated with oxaliplatin-based regimens with or without prior bevacizumab. While bevacizumab is a monoclonal antibody that binds vascular endothelial growth factor (VEGF)-A, aflibercept is a recombinant protein made of part of the VEGF receptors 1 and 2 fused to an immunoglobulin (Ig)-G1 Fc fragment. Improved mOS (HR 0.82, 95% CI 0.71–0.94, p = 0.0032) and mPFS (HR 0.76, 95% CI 0.66–0.87, p < 0.0001) was noted with FOLFIRI plus aflibercept compared with FOLFIRI in the second-line setting, and the findings did not vary depending on whether patients had received prior bevacizumab. 82
Ramucirumab, a humanized monoclonal antibody that binds the extracellular domain of VEGF receptor 2, has also been evaluated in combination with FOLFIRI after progression on first-line FOLFOX plus bevacizumab in the RAISE trial. mOS (HR 0.84, 95% CI 0.73–0.98, p = 0.022) and mPFS (HR 0.73, 95% CI 0.70–0.90, p = 0.0005) were improved compared with FOLFIRI plus placebo. 83 The benefit of second-line VEGF inhibition is statistically significant in all of these trials, however its clinical significance is unclear. An important note about second-line VEGF inhibition is that neither aflibercept nor ramucirumab have been compared with bevacizumab beyond progression. These novel agents represent a more expensive treatment option and the NCCN guidelines have endorsed preferential use of bevacizumab due to cost implications, however the benefit for all agents targeting angiogenesis beyond the first-line setting appears similar. 4
Third-line and fourth-line oral treatment options
Regorafenib and TAS-102 have been approved as new oral agents available for the treatment of patients with refractory mCRC. Regorafenib is a small molecule inhibitor with numerous targets, including VEGF receptors 1–3, platelet-derived growth factor receptor, tyrosine receptor kinase 2, fibroblast growth factor receptors, BRAF, KIT, and RET. In the phase III CORRECT trial, mOS improved from 5.0 months with placebo to 6.4 months with regorafenib at a preplanned interim analysis (HR 0.77, 95% CI 0.64–0.94, one-sided p = 0.0052). 11 The CONCUR trial was a confirmatory trial focusing on Asian patients. In CONCUR mOS was improved from 6.3 months with placebo to 8.8 months with regorafenib (HR 0.55, 95% CI 0.40–0.77, one-sided p = 0.00016). 84 In both trials there were significant rates of adverse events when patients were treated with regorafenib (93% and 97% respectively), with grade 3 or 4 toxicities occurring in over 50% of patients, of which hand–foot syndrome was the most common, occurring in 17% of patients. Similar toxicity results were obtained in the postmarketing trials CONSIGN and REBECCA.85,86 Trials assessing the ability of supportive interventions such as ginseng, exercise, and fish oil to mitigate fatigue [ClinicalTrials.gov identifier: NCT02581059, NCT02940223] and perindopril to decrease the incidence of hand–foot syndrome [ClinicalTrials.gov identifier: NCT02651415] are currently underway. A small unblinded study assessing the benefit of low-dose dexamethasone (2 mg/day orally) in combination with regorafenib suggested that dexamethasone may decrease treatment-related fatigue. 87
TAS-102 is a combination of trifluridine, a cytotoxic thymidine analog, and tipiracil, a thymidine phosphorylase inhibitor. In the RECOURSE clinical trial, TAS-102 increased mOS from 5.3 months to 7.1 months compared with placebo (HR 0.68, 95% CI 0.58–0.81, p < 0.001) in treatment-refractory mCRC. 10 Overall rate of adverse events was comparable between TAS-102 and placebo, however there were more grade 3 or 4 toxicities noted with TAS-102 (69% versus 52%). Most of the toxicities associated with TAS-102 are hematologic abnormalities, with grade 3 or 4 neutropenia occurring in 38% of patients, however rates of febrile neutropenia were low at 4%. Compared with regorafenib, a retrospective Japanese study suggested similar efficacy of the two agents regardless of sequence in which used. 88 The main difference between the agents was a higher rate of dose reductions with regorafenib due to adverse events. There were also higher rates of hand–foot syndrome and transaminitis with regorafenib, while TAS-102 was associated with more hematologic toxicity. At this point in time there is no evidence to suggest a preferred order for regorafenib and TAS-102 and patients in RECOURSE who had previously received regorafenib appeared to have similar benefit to those without prior regorafenib. 10
Future directions
Continued refinement in our understanding of the molecular subtypes of mCRC is essential to improve outcomes and identify patients who may benefit from individual targeted therapies. Tumor location has become an important factor guiding targeted therapy, however this is likely only a surrogate marker for molecular patterns that differ by location and that we do not yet understand. Several key mutations occurring in large proportions of mCRC have been identified, however our ability to study smaller subgroups will present a significant challenge. For example, the HERACLES trial had to screen 913 patients to identify 44 with HER2 amplifications. 65
Improvements in NGS assay characteristics and costs make targeted sequencing panels an ideal screening platform for clinical care. They can test multiple genes concurrently and can multiplex samples from different tumor histologies. This facilitates cost savings and deals with logistical barriers, such as delaying the run of a panel until a certain number of samples are available. The use of these panels is currently limited to defining RAS and BRAF mutation status outside of a clinical trial, but they will likely continue to expand.
Until now, the use of more comprehensive sequencing efforts such as whole exome sequencing (WES) or whole genome sequencing (WGS) have been limited to case reports of exceptional responders. 89 However, these technologies may play a greater role in the routine treatment of patients as sequencing costs fall and bioinformatic pipelines continue to improve. One of the major barriers to applying WES and WGS to clinical practice continues to be the lag between obtaining tissue and delivering clinically meaningful information to physicians. WES and WGS provide different information, with better identification of copy number alterations and structural rearrangement, but less depth to interrogate low-frequency mutations and tumor heterogeneity than panels. These two techniques may not be competing but complimentary assays that when combined with liquid biopsies represent a significant step forward in the treatment of CRC and allow interrogation of mechanisms of resistance and de-novo pathology. 90 There is an expanding range of tools available to clinicians and we must now learn how to use these tools to improve the outcomes for patients.
Footnotes
Acknowledgements
JL is a member of the UBC Clinician Investigator program and was the recipient of the CAMO 2016 Research Fellowship, the CCF of ASCO - J. Edward Mahoney Foundation YIA, and the RCPSC Detweiler Fellowship. The authors thank these programs for providing their support and making this work possible. JL performed this research in partial fulfillment of requirements for an MS degree from MD Anderson UTHealth GSBS. JL and SK were both responsible for conception and design, manuscript writing, and final approval of this manuscript.
Funding
SK is the recipient of NIH R01 grants that supported this research (CA 172670 and CA 187238).
Conflict of interest statement
The authors declare that there is no conflict of interest.